| |
Therapeutic Silencing of MicroRNA-122 by SPC3649 in Primates with Chronic Hepatitis C Virus Infection
|
| |
| |
Originally published in Science Express on 3 December 2009
Science 8 January 2010:
Vol. 327. no. 5962, pp. 198 - 201
DOI: 10.1126/science.1178178
Robert E. Lanford,1,* Elisabeth S. Hildebrandt-Eriksen,2,* Andreas Petri,2,* Robert Persson,2 Morten Lindow,2 Martin E. Munk,2 Sakari Kauppinen,2,3,* Henrik rum2,{dagger}
The liver-expressed microRNA-122 (miR-122) is essential for hepatitis C virus (HCV) RNA accumulation in cultured liver cells, but its potential as a target for antiviral intervention has not been assessed. We found that treatment of chronically infected chimpanzees with a locked nucleic acid (LNA)-modified oligonucleotide (SPC3649) complementary to miR-122 leads to long-lasting suppression of HCV viremia, with no evidence of viral resistance or side effects in the treated animals. Furthermore, transcriptome and histological analyses of liver biopsies demonstrated derepression of target mRNAs with miR-122 seed sites, down-regulation of interferon-regulated genes, and improvement of HCV-induced liver pathology. The prolonged virological response to SPC3649 treatment without HCV rebound holds promise of a new antiviral therapy with a high barrier to resistance.
1 Department of Virology and Immunology and Southwest National Primate Research Center, Southwest Foundation for Biomedical Research, San Antonio, TX 78227, USA.
2 Santaris Pharma, Kogle Alle 6, DK-2970 Hrsholm, Denmark.
3 Copenhagen Institute of Technology, Aalborg University, Lautrupvang 15, DK-2750 Ballerup, Denmark.
* These authors contributed equally to this work.
{dagger} To whom correspondence should be addressed. E-mail: hoe@santaris.com
Hepatitis C virus (HCV) infection is a leading cause of liver disease worldwide, with more than 170 million infected individuals at greatly increased risk of liver failure and hepatocellular carcinoma. The current standard anti-HCV therapy, which combines pegylated interferon-{alpha} (IFN-{alpha}) with ribavirin, provides sustained clearance of HCV in only about 50% of patients and is often associated with serious side effects (1, 2). Therapies that target essential host functions for HCV may provide a high barrier to resistance, and thus could present an effective approach for the development of new HCV antiviral drugs. MicroRNA-122 (miR-122) is a highly abundant, liver-expressed microRNA that binds to two closely spaced target sites in the 5' noncoding region (NCR) of the HCV genome, resulting in up-regulation of viral RNA levels (3, 4). Interaction of miR-122 with the HCV genome is essential for accumulation of viral RNA in cultured liver cells, and both target sites are required for modulation of HCV RNA abundance (3-5).
Previously, we reported on potent and specific miR-122 silencing in vivo using a locked nucleic acid (LNA)-modified phosphorothioate oligonucleotide (SPC3649) complementary to the 5' end of miR-122, which led to long-lasting decrease of serum cholesterol in mice and African green monkeys (6). Here, we investigated the potential of miR-122 antagonism by SPC3649 as a new anti-HCV therapy in chronically infected chimpanzees (genotype 1). Baseline measurements were obtained from four chimpanzees for 4 weeks, the last two of which included an intravenous (i.v.) placebo dose of saline. Two animals each were assigned to the high- and low-dose groups (5 mg kg-1 and 1 mg kg-1, respectively) and were treated with i.v. injections of SPC3649 on a weekly basis for 12 weeks (Fig. 1A), followed by a treatment-free period of 17 weeks. In the high-dose group, a significant decline of HCV RNA in the serum was detected 3 weeks after the onset of SPC3649 dosing, with a maximum decrease of 2.6 orders of magnitude in HCV RNA levels 2 weeks after end of treatment (Fig. 1A). Analysis of HCV RNA levels in the liver showed a decrease of 2.3 orders of magnitude in the high-dose animals. One low-dose animal achieved a viral decline of 1.3 orders of magnitude; the other experienced fluctuations in HCV RNA levels during dosing that made evaluation of the degree of suppression difficult (Fig. 1A).
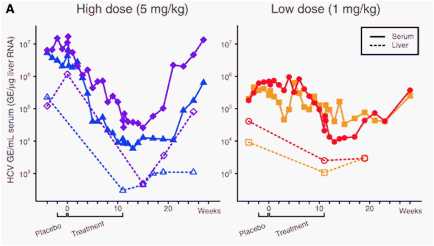
We next assessed the in vivo antagonism of miR-122 in chimpanzee liver biopsies. Mature miR-122 was detected in the baseline samples (week -4) from all animals, whereas SPC3649 was detected in RNA samples obtained during treatment and up to 8 weeks after the last dose in the high-dose animals. This coincided with sequestration of miR-122 in a heteroduplex with SPC3649, as detected by a shifted band on Northern blots (Fig. 1B) (6). Quantification of miR-122 levels by real-time reverse transcription polymerase chain reaction showed a factor of >300 decrease in free miR-122 levels for the high-dose animals (fig. S1). Free SPC3649 was markedly reduced in the liver at week 25 in the high-dose animals, accompanied with detection of free miR-122 alongside the miR-122:SPC3649 heteroduplex band (Fig. 1B). These findings demonstrate efficient delivery of SPC3649 to the chimpanzee liver, resulting in potent and sustained antagonism of miR-122. The reason for the reduced response in the low-dose group was not apparent from the Northern data, because no miR-122 was detected at the end of dosing. It is possible that low levels of miR-122 undetectable by Northern blot could still be sufficient to support HCV RNA accumulation in these animals. A recent report on markedly decreased miR-122 levels in interferon nonresponder patients with chronic HCV infection supports the notion that even low levels of miR-122 could facilitate HCV abundance in vivo (7).
Two lines of evidence imply that no viral resistance to therapy occurred during treatment with SPC3649. First, no rebound in viremia was observed during the 12-week dosing phase; HCV RNA levels were still an order of magnitude below baseline 16 weeks after dosing (4x0513, Fig. 1A). Second, deep sequencing of the HCV 5' NCR from the two high-dose animals-which yielded between 73,000 and 214,000 reads each at four time points (baseline, end of treatment, viral rebound, and end of the follow-up period)-showed no enrichment of adaptive mutations in the miR-122 seed sites (Fig. 1C and fig. S2). This is consistent with the fact that both miR-122 sites are conserved in all HCV genotypes and subtypes (Fig. 1D and figs. S3 to S9). The lack of viral resistance during SPC3649 therapy is in stark contrast to what has been observed with direct acting antivirals in HCV-infected chimpanzees. Within 2 days of dosing with a non-nucleoside polymerase inhibitor, 67% of the HCV clones already possessed known resistance mutations, with 10% of the clones having two resistance mutations, which triggered a rapid rebound in viremia (8).
We next investigated the effect of miR-122 antagonism on the chimpanzee liver transcriptome by expression profiling of the liver biopsies performed after SPC3649 treatment relative to baseline samples. Liver mRNAs with miR-122 seed match sites in the 3' untranslated regions (UTRs) showed a significant tendency to be derepressed in both high-dose animals and the responding low-dose animal relative to transcripts without miR-122 sites (Fig. 2A, P = 7.3 x 10-4, 1.0 x 10-3, and 2.2 x 10-10 for 8-mer seed sites, respectively, Kolmogorov-Smirnov test; see also fig. S10). A total of 259 mRNAs with 8-mer miR-122 seed sites were identified by this approach (table S1). By contrast, no significant target mRNA derepression was observed in the weakly responding low-dose animal (Fig. 2A). We also examined the expression data for changes related to prolonged decrease in viral RNA during SPC3649 therapy. A supervised analysis of chimpanzee interferon-regulated genes (IRGs) (9, 10) revealed that the reduction in viremia was clearly associated with down-regulation of most IRGs in the high-dose animals and the responding low-dose animal (Fig. 2B and table S2). This correlated with the measured serum levels of the chemokine IP-10 (CXCL10), a highly induced IRG in HCV infections, which thereby provides a readily accessible biomarker of the hepatic IFN response during SPC3649 therapy (Fig. 2C). Together, these data imply that the endogenous IFN pathway in the liver is rapidly normalized in response to inhibition of HCV RNA accumulation even when therapy does not completely eradicate detectable viral RNA. Nonresponders to IFN-{alpha}-based HCV therapy have increased hepatic levels of IRG transcripts and serum IP-10 protein levels (11-17), reflecting a maximally induced and nonresponsive hepatic IFN response. The chimpanzee appears to be an extreme representative of this phenotype in human HCV patients, designated as IFN null-responders (18). Thus, our finding that treatment with SPC3649 results in normalization of IRG levels suggests that this therapy could be used to convert IFN null-responders to responders by reducing the viral load, thereby permitting the endogenous IFN pathway to reset to responsiveness.
Antagonism of miR-122 in chimpanzees by SPC3649 led to markedly lowered serum cholesterol in the high-dose group (Fig. 2D), similar to observations in mice and in African green monkeys (6, 19). One of the high-dose animals had a maximum decline of 44% at week 14, whereas the other animal showed a 29% decrease in cholesterol at the same time point. Pronounced decreases were observed in both low-density lipoprotein (LDL) (25 to 54%) and apolipoprotein apo-B, its primary lipoprotein component (23 to 42%) (fig. S12). In contrast to our previous findings in monkeys, where decreases in high-density lipoprotein (HDL) and its major apolipoprotein apo-A1 were more pronounced relative to LDL and apo-B (7), the observed changes in HDL or apo-A1 in chimpanzees were more variable (fig. S13). Thus, it is possible that the cholesterol-lowering effect of miR-122 antagonism is different in chimpanzees and may better reflect the expected response in humans.
To assess the safety of miR-122 antagonism after prolonged treatment with SPC3649, we monitored an extensive set of clinical chemistries and correlated them with plasma levels of the compound. The peak plasma concentrations (Cmax) were dose-proportional and similar after first and last dose, ranging from 6.1 to 6.3 µg/ml for the low-dose animals and from 17.7 to 30.6 µg/ml for the high-dose animals (Table 1). The terminal plasma half-life was about 20 days in the high-dose animals. The plasma trough levels at the high dose ranged from 31 to 67 ng/ml and were maintained at this level for 4 weeks after the last dose (Fig. 3A). Complete blood counts, blood chemistries, coagulation markers, urinalysis, and complement activation were determined throughout the study, as were lymphocyte subsets, circulating cytokine-chemokine profiles, and additional safety parameters (table S3). No SPC3649-related abnormalities were observed for any of the measurements (Fig. 3, B and C, figs. S14 and S15, and table S3). A spike in alanine aminotransferase (ALT) was observed in one high-dose animal (4x0514), but this commenced prior to the first dose and resolved in the early dosing phase (Fig. 3B). Notably, during therapy ALT was reduced to normal levels, likely due to reduction in the viral load, and was again elevated at the end of the follow-up period when viremia returned to baseline. Histology examinations of the baseline liver biopsies from the high-dose animals revealed HCV-specific changes, including mild hepatocellular swelling with disruption of hepatocellular sinuses and cords (Fig. 3, D to G, and fig. S16). Improved liver histology was observed in both high-dose animals after treatment at week 19, indicating a response to prolonged suppression of viremia and normalization of the IFN pathway.
Our results show that antagonism of miR-122 by the LNA oligonucleotide SPC3649 leads to marked suppression of viremia in chronically HCV-infected chimpanzees, thus implying that miR-122 is essential for accumulation of HCV RNA in vivo. The good PK properties, safety profile, and high stability of SPC3649 in vivo, combined with the prolonged suppression of viremia beyond treatment, suggest that less frequent dosing could be used after viral suppression is attained. SPC3649 therapy provided a high barrier to resistance, as shown by the lack of rebound in viremia during the 12-week treatment and the lack of adaptive mutations in the two miR-122 seed sites of HCV 5' NCR. Conservation of both miR-122 seed sites in all HCV genotypes and subtypes suggests that such therapy will be genotype-independent. Finally, this study demonstrates the feasibility and safety of prolonged administration of a LNA oligonucleotide drug that antagonizes the function of a specific microRNA in a highly relevant disease model.
|
|
| |
| |
|
|
|