| |
Telaprevir with Peginterferon and Ribavirin for Chronic HCV Genotype 1 Infection...PROVE1 in NEJM 4/2009
|
| |
| |
Download the PDF here
NEJM April 30 2009
John G. McHutchison, M.D., Gregory T. Everson, M.D., Stuart C. Gordon, M.D., Ira M. Jacobson, M.D., Mark Sulkowski, M.D., Robert Kauffman, M.D., Lindsay McNair, M.D., John Alam, M.D., Andrew J. Muir, M.D., for the PROVE1 Study Team
"The rate of sustained virologic response was 41% (31 of 75 patients) in the PR48 group, as compared with 61% (48 of 79 patients) in the T12PR24 group (P=0.02), 67% (53 of 79 patients) in the T12PR48 group (P=0.002), and 35% (6 of 17 patients) in the T12PR12 group (this group was exploratory and not compared with the control group). Viral breakthrough occurred in 7% of patients receiving telaprevir. The rate of discontinuation because of adverse events was higher in the three telaprevir-based groups (21%, vs. 11% in the PR48 group), with rash the most common reason for discontinuation....
....Patients were enrolled at 37 centers in the United States. Eligible patients were 18 to 65 years of age, had a chronic genotype 1 HCV infection, and had not been treated previously for hepatitis C. This was a phase 2b, randomized, parallel-group, double-blind, placebo-controlled trial. Patients who completed the screening were stratified according to self-reported race or ethnic group (black vs. any other) and baseline weight (>75 kg vs. <75 kg) and were randomly assigned to one of four treatment groups....
....addition of a hepatitis CĐspecific protease inhibitor, telaprevir, to current standard therapy consisting of peginterferon alfa-2a and ribavirin can significantly improve the rate of sustained virologic response in patients infected with HCV genotype 1, as compared with the rate with standard therapy. The observed sustained virologic response in the PR48 group (41%) is consistent with previous reports.....duration of therapy can be reduced from 48 weeks to 24 weeks for most patients while maintaining an improved sustained virologic response....The overall responses reflect a marked increase in the rate of rapid virologic response at week 4, and a low subsequent rate of relapse, with telaprevir-based treatment as compared with standard therapy. With the addition of peginterferon alfa-2a and ribavirin to the telaprevir-based regimen, viral breakthrough was infrequent (seen in 7% of patients). Although there were relatively few black patients treated in this study, the rate of sustained virologic response of 44% among black patients in the telaprevir-based groups is encouraging.....The relapse rates that we observed for the 24-week and 48-week treatment regimens that included telaprevir (2% and 6%, respectively) were lower than the relapse rate observed in the control group (23%); the rate in the control group was within the range of the reported rates of 18 to 30% for current standard HCV antiviral therapy.5,20 Moreover, the relapse rate of 2% in the T12PR24 group indicates that a 24-week treatment duration is sufficient in patients who have a rapid virologic response. Patients randomly assigned to the T12PR24 group who did not have a rapid virologic response received peginterferon and ribavirin for an extended period Ń 48 weeks. Therefore, this study cannot evaluate whether 24-week treatment would be sufficient in this subgroup.....The addition of telaprevir to therapy with peginterferon alfa-2a and ribavirin was associated with an increase in the rate of treatment discontinuation, predominantly owing to the side effect of rash. In a previous study of telaprevir and peginterferon alfa-2a, rash was more common in the two groups receiving telaprevir than in the group receiving peginterferon alfa-2a alone....
....The most common adverse events were consistent with typical interferon-induced systemic symptoms (Table 3). However, certain adverse events Ń such as rash, pruritus, nausea, and diarrhea Ń were more common in the groups that received telaprevir. The proportion of patients who discontinued treatment because of an adverse event was higher in the three telaprevir-based treatment groups (21%) than in the PR48 group (11%) (Table 4). Serious adverse events were reported in 22 patients during treatment (4 patients in the PR48 group and 18 patients in the telaprevir-based groups). Fifteen of the serious adverse events were considered to be related to the study drug regimen. Serious adverse events in more than one patient, which occurred only in the telaprevir-based groups, included rash (three patients), anemia (three patients), ocular events (retinal detachment and scotoma, three patients), and depression (two patients).....
.....Rash was more common in the telaprevir-based groups than in the control group, with mild rash occurring in 37% and 32% of patients, respectively; moderate rash, in 15% and 8%, respectively; and severe rash, in 7% and 1%, respectively. The rashes were described as typical drug-induced maculopapular rashes. Pruritus was sometimes, but not always, considered to be associated with rash. The site investigators determined the severity of the rashes. When it became clear that there was an increased incidence of rash, the protocol was amended to include guidance for the grading of rash, but few rashes occurred after that time. The protocol did not provide specific guidance for discontinuation of treatment because of rash, although in most cases of severe rash, one or more of the study drugs was stopped. The median time to treatment discontinuation because of rash in the telaprevir groups was 73 days (range, 8 to 88) after the start of the study treatment. All cases of severe rash, which were followed by the site investigators and medical monitors, resolved after the discontinuation of treatment.....
.....A decrease in hemoglobin levels was more common in patients receiving telaprevir-based regimens than in controls during the first 12 weeks of treatment. Erythropoietin use was prohibited during the first 12 weeks of treatment to allow for accurate assessment of any potential effect on hemoglobin levels. Patients in the PR48 group had a median decline in the hemoglobin level of about 3 g per deciliter at week 12; the decline in the telaprevir groups was 0.5 to 1 g per deciliter greater. After telaprevir was discontinued, the hemoglobin values increased to the mean level in the control group and remained similar to control levels thereafter."
ABSTRACT
Background- Current therapy for chronic hepatitis C virus (HCV) infection is effective in less than 50% of patients infected with HCV genotype 1. Telaprevir, a protease inhibitor specific to the HCV nonstructural 3/4A serine protease, rapidly reduced HCV RNA levels in early studies.
Methods- We randomly assigned patients infected with HCV genotype 1 to one of three telaprevir groups or to the control group. The control group (called the PR48 group) received peginterferon alfa-2a (180 µg per week) and ribavirin (1000 or 1200 mg per day, according to body weight) for 48 weeks, plus telaprevir-matched placebo for the first 12 weeks (75 patients). The telaprevir groups received telaprevir (1250 mg on day 1 and 750 mg every 8 hours thereafter) for 12 weeks, as well as peginterferon alfa-2a and ribavirin (at the same doses as in the PR48 group) for the same 12 weeks (the T12PR12 group, 17 patients) or for a total of 24 weeks (the T12PR24 group, 79 patients) or 48 weeks (the T12PR48 group, 79 patients). The primary outcome was a sustained virologic response (an undetectable HCV RNA level 24 weeks after the end of therapy).
Results- The rate of sustained virologic response was 41% (31 of 75 patients) in the PR48 group, as compared with 61% (48 of 79 patients) in the T12PR24 group (P=0.02), 67% (53 of 79 patients) in the T12PR48 group (P=0.002), and 35% (6 of 17 patients) in the T12PR12 group (this group was exploratory and not compared with the control group). Viral breakthrough occurred in 7% of patients receiving telaprevir. The rate of discontinuation because of adverse events was higher in the three telaprevir-based groups (21%, vs. 11% in the PR48 group), with rash the most common reason for discontinuation.
Conclusions- Treatment with a telaprevir-based regimen significantly improved sustained virologic response rates in patients with genotype 1 HCV, albeit with higher rates of discontinuation because of adverse events. (ClinicalTrials.gov number, NCT00336479 [ClinicalTrials.gov] .)
Worldwide, an estimated 180 million people have a chronic infection with hepatitis C virus (HCV).1 Hepatitis C is a leading cause of cirrhosis and hepatocellular carcinoma and is the leading indication for liver transplantation in the United States.2,3 The current treatment for HCV infection is peginterferon alfa combined with ribavirin and administered for 24 weeks (for HCV genotype 2 or 3) or 48 weeks (for HCV genotype 1, the most prevalent genotype in Europe and North America).4 The aim of HCV therapy is a sustained virologic response, defined as an undetectable serum HCV RNA level 24 weeks after cessation of therapy. For patients with HCV genotype 1, the rate of sustained virologic response ranges between 38 and 46%.5,6,7 In subgroups of this population, the rate of sustained virologic response is even lower, including among black patients, who have a reported rate of sustained virologic response of 19%.8
Efforts to improve patients' outcomes have focused on antiviral therapy specifically targeted to HCV, and several agents in development inhibit either the HCV polymerase or protease.9,10 Telaprevir is an orally bioavailable inhibitor of the nonstructural 3/4A (NS3/4A) HCV serine protease.11 In phase 1b trials, telaprevir monotherapy for 14 days resulted in a reduction of up to 5 log10 units in plasma HCV RNA levels in subjects infected with HCV genotype 1.12 However, some subjects had evidence of viral breakthrough, and viral sequence analysis revealed resistance-associated mutations in the catalytic domain of the NS3 protease.13 Data from subsequent phase 1b studies suggested that combining telaprevir with peginterferon, with or without ribavirin, increased viral inhibition and decreased the emergence of resistance.14,15,16
Protease Inhibition for Viral Evaluation 1 (PROVE1) was a phase 2b, randomized, double-blind, multicenter study of telaprevir in combination with peginterferon alfa-2a and ribavirin in patients infected with HCV genotype 1 in the United States who had not previously been treated. We designed the study to assess the safety and efficacy of telaprevir-based therapy and to explore whether this agent could shorten the duration of the current standard therapy.
Results
Characteristics of the Patients
Between June 2006 and September 2006, a total of 263 patients underwent randomization, and 250 received at least one dose of a study drug (Figure 1). The baseline characteristics were similar among the treatment groups (Table 1). The majority of patients (157 of 250 [63%]) were men, and the mean age was 48.1 years. Whites accounted for 77% of the study population (192 of 250 patients). Most patients (218 of 250 [87%]) had HCV RNA levels of 800,000 IU or more per milliliter at baseline.
Efficacy
The sustained virologic response rate was 61% (48 of 79 patients) in the T12PR24 group, as compared with 41% (31 of 75 patients) in the PR48 group (P=0.02) (Table 2). Rates of sustained virologic response were 67% (53 of 79 patients) in the T12PR48 group (P=0.002 and P=0.51 for the comparison with the PR48 group and the T12PR24 group, respectively) and 35% (6 of 17 patients) in the T12PR12 group (Table 2). In the small subgroup of black patients enrolled in the study, rates of sustained virologic response were 11% (1 of 9 patients) in the PR48 group and 44% (8 of 18 patients) in the telaprevir-based groups.
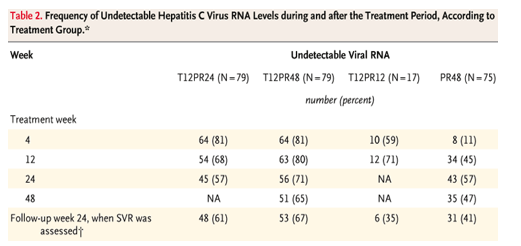
Rates of rapid virologic response (undetectable HCV RNA levels at week 4) were higher with telaprevir-based therapy than without it (in the PR48 group) (P<0.001 for each comparison) (Table 2 and Figure 2). In the T12PR24 and T12PR12 groups, patients who had neither a rapid virologic response nor HCV RNA levels that remained undetectable through week 20 (for T12PR24) or through week 10 (for T12PR12), were to continue treatment with peginterferon and ribavirin for 48 weeks. Of the 15 patients who were to continue treatment, 9 discontinued therapy before it was completed and 1, in the T12PR24 group, completed the 48 weeks of treatment and had a sustained virologic response (Figure 1). In the analyses, this patient was counted as not having had a response.
At baseline, 20% of the patients in the PR48 group and 25% of the patients in the telaprevir-based groups had alanine aminotransferase values within the normal range. At the end of the treatment period, 75% of patients in the PR48 group and 76% of those in the telaprevir-based groups had normal alanine aminotransferase values.
Only 1 of 41 patients (2%) in the T12PR24 group had a relapse (undetectable HCV RNA at the time of completion of treatment but detectable levels during the follow-up period), whereas 3 of 51 patients (6%) in the T12PR48 group and 3 of 9 patients (33%) in the T12PR12 group had a relapse. In the PR48 group, 8 of 35 patients (23%) had a relapse.
Among the telaprevir-treated patients, 7% (12 of 175) had viral breakthrough (an increase of >1 log10 unit of HCV RNA as compared with the lowest value during the treatment period, or if the HCV RNA level had become undetectable, an increase to an HCV RNA value of >100 IU per milliliter). Most breakthroughs occurred during weeks 1 through 4 (in 9 of 12 patients), with the remainder (in 3 patients) occurring between weeks 5 and 12. Ten of the 12 patients had breakthroughs before the HCV RNA level became undetectable. The viral population at the time of breakthrough consisted predominantly of variants containing mutations V36M and R155K (in 10 patients infected with HCV genotype 1a) or A156T (in 1 patient with HCV genotype 1b). The remaining patient had missed several days of dosing and had a breakthrough at day 8; this patient was infected with predominantly wild-type virus.
Safety
The most common adverse events were consistent with typical interferon-induced systemic symptoms (Table 3). However, certain adverse events Ń such as rash, pruritus, nausea, and diarrhea Ń were more common in the groups that received telaprevir. The proportion of patients who discontinued treatment because of an adverse event was higher in the three telaprevir-based treatment groups (21%) than in the PR48 group (11%) (Table 4). Serious adverse events were reported in 22 patients during treatment (4 patients in the PR48 group and 18 patients in the telaprevir-based groups). Fifteen of the serious adverse events were considered to be related to the study drug regimen. Serious adverse events in more than one patient, which occurred only in the telaprevir-based groups, included rash (three patients), anemia (three patients), ocular events (retinal detachment and scotoma, three patients), and depression (two patients).
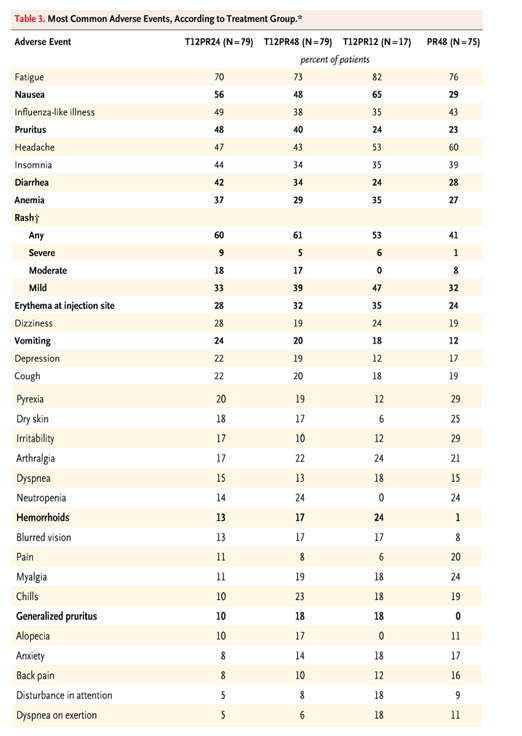
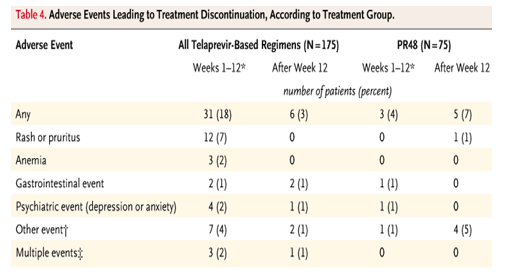
Rash was more common in the telaprevir-based groups than in the control group, with mild rash occurring in 37% and 32% of patients, respectively; moderate rash, in 15% and 8%, respectively; and severe rash, in 7% and 1%, respectively. The rashes were described as typical drug-induced maculopapular rashes. Pruritus was sometimes, but not always, considered to be associated with rash. The site investigators determined the severity of the rashes. When it became clear that there was an increased incidence of rash, the protocol was amended to include guidance for the grading of rash, but few rashes occurred after that time. The protocol did not provide specific guidance for discontinuation of treatment because of rash, although in most cases of severe rash, one or more of the study drugs was stopped. The median time to treatment discontinuation because of rash in the telaprevir groups was 73 days (range, 8 to 88) after the start of the study treatment. All cases of severe rash, which were followed by the site investigators and medical monitors, resolved after the discontinuation of treatment.
Changes in laboratory values during the study were consistent with those reported in association with the combined use of peginterferon and ribavirin.6,18 A decrease in hemoglobin levels was more common in patients receiving telaprevir-based regimens than in controls during the first 12 weeks of treatment. Erythropoietin use was prohibited during the first 12 weeks of treatment to allow for accurate assessment of any potential effect on hemoglobin levels. Patients in the PR48 group had a median decline in the hemoglobin level of about 3 g per deciliter at week 12; the decline in the telaprevir groups was 0.5 to 1 g per deciliter greater. After telaprevir was discontinued, the hemoglobin values increased to the mean level in the control group and remained similar to control levels thereafter. No other clinically significant differences in laboratory abnormalities were noted between the telaprevir groups and the control group.
Discussion
The results of this study, as well as those of a similar study conducted in Europe (the PROVE2 study, reported elsewhere in this issue of the Journal),19 suggest that the addition of a hepatitis CĐspecific protease inhibitor, telaprevir, to current standard therapy consisting of peginterferon alfa-2a and ribavirin can significantly improve the rate of sustained virologic response in patients infected with HCV genotype 1, as compared with the rate with standard therapy. The observed sustained virologic response in the PR48 group (41%) is consistent with previous reports.5,6,7 Our study also shows that the duration of therapy can be reduced from 48 weeks to 24 weeks for most patients while maintaining an improved sustained virologic response. The overall responses reflect a marked increase in the rate of rapid virologic response at week 4, and a low subsequent rate of relapse, with telaprevir-based treatment as compared with standard therapy. Although there were relatively few black patients treated in this study, the rate of sustained virologic response of 44% among black patients in the telaprevir-based groups is encouraging.
The relapse rates that we observed for the 24-week and 48-week treatment regimens that included telaprevir (2% and 6%, respectively) were lower than the relapse rate observed in the control group (23%); the rate in the control group was within the range of the reported rates of 18 to 30% for current standard HCV antiviral therapy.5,20 Moreover, the relapse rate of 2% in the T12PR24 group indicates that a 24-week treatment duration is sufficient in patients who have a rapid virologic response. Patients randomly assigned to the T12PR24 group who did not have a rapid virologic response received peginterferon and ribavirin for an extended period Ń 48 weeks. Therefore, this study cannot evaluate whether 24-week treatment would be sufficient in this subgroup.
The addition of telaprevir to therapy with peginterferon alfa-2a and ribavirin was associated with an increase in the rate of treatment discontinuation, predominantly owing to the side effect of rash. In a previous study of telaprevir and peginterferon alfa-2a, rash was more common in the two groups receiving telaprevir than in the group receiving peginterferon alfa-2a alone.15 Guidelines for the management of rash have been incorporated in all subsequent clinical protocols involving telaprevir; the guidelines were added to this protocol, but almost all patients had completed the telaprevir-dosing period by that time, so the effect on the outcome in this study cannot be assessed. The exact mechanism of action leading to rash in patients receiving telaprevir remains unknown. An adverse effect on hemoglobin was also noted with telaprevir-based regimens; this effect was of low magnitude and was reversed within 4 weeks after telaprevir dosing ended.
This study was designed to evaluate telaprevir in combination with peginterferon alfa-2a and ribavirin in a treatment regimen with regard to the rate of sustained virologic response and to compare telaprevir-based treatment with current therapy. With the addition of peginterferon alfa-2a and ribavirin to the telaprevir-based regimen, viral breakthrough was infrequent (seen in 7% of patients). Viral variants observed in patients at the time of breakthrough in this study carried telaprevir-resistanceĐassociated mutations similar to those described in studies of shorter duration.16 These variants are sensitive to interferon and ribavirin in vitro21 and have been shown previously to respond to subsequent interferon and ribavirin treatment in some patients.16 Theoretically, the patients with viral breakthrough may have had a reduced response to peginterferon and ribavirin.13,16 The potential long-term clinical consequences of the selection and persistence of these variants in patients who do not achieve a sustained virologic response are unknown and will require long-term follow-up studies.
In conclusion, the addition of telaprevir to peginterferon alfa-2a and ribavirin in patients infected with hepatitis C genotype 1 who had not been treated previously significantly increased the rate of sustained virologic response; this approach may allow for a substantial reduction in the duration of therapy in most patients.
Methods
Subjects
Patients were enrolled at 37 centers in the United States. Eligible patients were 18 to 65 years of age, had a chronic genotype 1 HCV infection, and had not been treated previously for hepatitis C. Patients were also seronegative for hepatitis B surface antigen and antibodies against human immunodeficiency virus types 1 and 2, and had an absolute neutrophil count of 1500 or more per cubic millimeter, a platelet count of 90,000 or more per cubic millimeter, and a normal hemoglobin level. Patients were excluded if they had decompensated liver disease, another cause of clinically significant liver disease, hepatocellular carcinoma, or histologic evidence of cirrhosis (on liver biopsy, which was required within 2 years before the study). The protocol was approved by independent or institutional review boards of all study centers, and all patients provided written informed consent before participating in the study.
Study Design and Organization
This was a phase 2b, randomized, parallel-group, double-blind, placebo-controlled trial. Patients who completed the screening were stratified according to self-reported race or ethnic group (black vs. any other) and baseline weight (>75 kg vs. 75 kg) and were randomly assigned to one of four treatment groups. The T12PR24 group received telaprevir (VX-950, Vertex Pharmaceuticals) plus peginterferon alfa-2a (Pegasys, Roche) and ribavirin (Copegus, Roche) for 12 weeks, followed by peginterferon alfa-2a and ribavirin for 12 more weeks. The T12PR48 group received telaprevir plus peginterferon alfa-2a and ribavirin for 12 weeks, followed by peginterferon alfa-2a and ribavirin for 36 more weeks. The T12PR12 group received telaprevir plus peginterferon alfa-2a and ribavirin for 12 weeks. The PR48 (control) group received placebo plus peginterferon alfa-2a plus ribavirin for 12 weeks, followed by peginterferon alfa-2a and ribavirin for 36 more weeks.
Telaprevir was given at a dose of 1250 mg on day 1, followed by a dose of 750 mg every 8 hours; peginterferon alfa-2a was given at a dose of 180 µg per week by subcutaneous injection; and ribavirin was given orally, at a dose of 1000 mg per day (for body weight <75 kg) or 1200 mg per day (for body weight >75 kg). Because this was the first study exploring shorter-than-standard durations of therapy (<48 weeks), before initiation of the study it was planned that only 20 patients would be randomly assigned to the group with the shortest treatment period (T12PR12). For the first 80 patients, randomization was to be performed at a 1:1:1:1 ratio among the four groups (with randomization blocks of 4). The remaining 180 patients were to be randomized in a 1:1:1 ratio among the T12PR24, T12PR48, and PR48 groups (with randomization blocks of 3).
As is consistent with current standard practice,6,17 therapy was discontinued in patients in the PR48 group who did not have a decline of 2 log10 units in the HCV RNA level by week 12 or undetectable HCV RNA levels at week 24. Patients in the T12PR24 and T12PR12 groups were required to have undetectable HCV RNA levels by week 4 (known as a rapid virologic response). If these patients had detectable HCV RNA at any time during weeks 4 through 20 (for T12PR24) or weeks 4 through 10 (for T12PR12), they were not permitted to undergo the assigned duration of therapy (12 or 24 weeks) and instead continued to receive peginterferon alfa-2a and ribavirin for the standard total of 48 weeks. This approach was based on the hypothesis that for patients with a slow virologic response, longer treatment with peginterferon alfa-2a would increase the likelihood of a sustained response. Because the intention was to assess the response to shorter-than-standard durations of therapy, if patients in the T12PR24 and T12PR12 groups underwent treatment for a longer period than the assigned duration, they were considered to have treatment failure in the analysis.
The study sponsor and two academic principal investigators were jointly responsible for study design and protocol development. All authors had access to the data, assume responsibility for the accuracy and completeness of the data reported, and contributed to the writing of the manuscript.
Efficacy Assessments
Plasma HCV RNA levels were measured with the use of the COBAS TaqMan HCV assay, version 1.0 (Roche Molecular Systems), with a lower limit of quantification of 30 IU per milliliter and a lower limit of detection of 10 IU per milliliter. Study visits, at which HCV RNA levels were measured, occurred at the time of screening and during the treatment period on days 1 and 4 and at weeks 1, 2, 3, 4, 6, 8, 10, 12, 16, 20, 24, 28, 36, and 48. Patients in the T12PR24 and T12PR12 groups completed visits at week 24 or 12 of the treatment period, respectively.
Patients had a safety follow-up visit 2 weeks after the completion of treatment (whether it was early or as planned). Patients who had undetectable HCV RNA levels at the time of completion of treatment had follow-up visits 4, 12, and 24 weeks afterward, at which time HCV RNA levels were measured. Patients who had detectable HCV RNA levels were not required to have further follow-up.
Safety Assessments
Chemical and hematologic assessments were performed at each study visit during the treatment period and at the safety follow-up visit. At each visit, data on adverse events were collected and physical examinations were performed, if clinically indicated.
Viral Breakthrough and Evaluation of HCV Sequence
Patients were followed for viral breakthrough during the first 12 weeks of treatment. Breakthrough was defined as an increase in the HCV RNA level of 1 log10 unit, as compared with the lowest value, or as an increase to an HCV RNA value of more than 100 IU per milliliter, if the HCV RNA had become undetectable. Patients who had viral breakthrough discontinued telaprevir or placebo but continued to receive peginterferon alfa-2a plus ribavirin for up to 48 weeks.
Blood samples were collected for viral sequencing at every study visit. Among patients who had viral breakthrough, for all samples for which the HCV RNA level was greater than 1000 IU per milliliter (the limit of detection of the sequencing assay), the NS3/4A region of the HCV genome was analyzed by means of population sequencing.
Rates of relapse during the follow-up period were calculated for the patients in each group who had undetectable HCV RNA at the time at which their assigned treatment was completed (24 weeks for the T12PR24 group, 12 weeks for the T12PR12 group, and 48 weeks for the T12PR48 and PR48 groups). In the T12PR24 and T12PR12 groups, relapse rates were based on data from the patients who had a rapid virologic response at treatment week 4 and undetectable HCV RNA levels through week 20 (for T12PR24) or through week 10 (for T12PR12) and who completed therapy.
Statistical Analysis
The primary end point was the proportion of patients in each group who had undetectable plasma HCV RNA levels 24 weeks after the completion of therapy (sustained virologic response). Analyses of efficacy and safety included data from all patients who had undergone randomization and had received at least one dose of any study drug. The primary planned analysis, on which the sample size was based, was the comparison of the PR48 group and the T12PR24 group. This analysis assumed a 50% response rate in the PR48 group and a 75% response rate in the T12PR24 group, with the use of a two-sided t-test, a significance level of 5%, and a sample of 80 patients who could be evaluated per group, to provide a statistical power of at least 85% to show a significant difference. The T12PR12 group was intentionally smaller than the other three groups, for an exploratory assessment of a very short duration of therapy; the study-analysis plan did not include a comparison of this group with the PR48 group.
Four planned interim analyses of safety and efficacy were conducted during the study at prespecified milestones, as was one analysis of safety only, when 80 patients had completed 12 weeks of treatment. Data management and interim analyses were performed by the Duke Clinical Research Institute; after the study was unblinded, data analyses were performed by Vertex Pharmaceuticals, the sponsor. An independent data monitoring committee reviewed the results of all interim analyses. The committee made recommendations regarding the study, but there were no predefined study-stopping rules. The primary outcome of the study was a sustained virologic response. Comparison of the primary end point between the control and telaprevir-based groups was not possible in any analysis except the final analysis, because the controls had not reached the time point at which the rate of sustained virologic response was to be calculated. Therefore, adjustments of the P value for the interim analyses were not necessary. All reported P values are two-sided.
|
|
| |
| |
|
|
|