| |
Investigational HIV agents for salvage
|
| |
| |
Pozniak, Anton L
HIV Services, St Stephen's Centre, Chelsea and Westminster Hospital NHS Foundation Trust, London, UK
Correspondence to Dr Anton L. Pozniak, MD, FRCP, Consultant Physician and Director of HIV Services, St Stephen's Centre, Chelsea and Westminster Hospital NHS Foundation Trust, 369 Fulham Road, London UK Tel: +44 2087465620; fax: +44 2088468299; e-mail: anton.pozniak@chelwest.nhs.uk
Current Opinion in HIV and AIDS:
November 2009 - Volume 4 - Issue 6 - p 524-530
doi: 10.1097/COH.0b013e328331d56f
Salvage therapy: Edited by Christine Katlama and Paul Sax
from Jules: I think there are additional efforts in development of investigational new HIV drugs not discussed here. Here is study presented at the 2009 Resistance Workshop in Ft Myers Florida about a new nucleotide from Gilead:
The HIV-1 RT Mutant Q151L Shows Decreased Replication Capacity, Selective High-Level Resistance to GS-9148 and Hypersusceptibility to Tenofovir and Zidovudine - (06/15/09)
Abstract
Purpose of review: There have been several new compounds developed from new and old antiretroviral drug classes recently that are active against highly resistant HIV-1. The management of patients who are highly drug experienced is complex and challenging and this article reviews current knowledge on the investigational agents in development for these patients. This review will examine some of the compounds that may have a role to play in highly experienced patients in the future.
Recent findings: Studies in early phases of therapy and some dose-ranging efficacy and safety clinical trials of investigational drugs have brought some drugs closer to licensing. New drugs in established classes not only have to be less toxic than the already licensed compounds but also have to be active against resistant viruses, these conditions make drug development difficult.
Summary: It is now possible for the majority of patients who are highly experienced in terms of drug treatment and who have virological failure to achieve HIV-1 viral loads below the limit of detection. There are, however, some patients who continue to have virological failure and develop further resistance and these patients need therapy that is effective; new drugs are being developed to meet this need.
Introduction
With the development of boosted protease inhibitors that had efficacy against viruses with multiple protease mutations, a new non nucleoside reverse transcriptase inhibitor (NNRTI) that is effective against some viruses with various NNRTI's mutations and two new classes of drugs (the entry/fusion and integrase inhibitors) salvage therapy needs to be redefined. The term salvage, in spite of its pejorative tone, still lingers on but has been replaced in most cases by the term 'highly treatment experienced'. Such patients, in terms of treatment options, can be divided into those in whom an effective suppressive regime can be constructed and those for whom an effective regimen cannot. These latter patients are the new 'salvage'. Many of these drugs adhere poorly but others have very limited antiviral options and require new therapies. These new compounds have to be active against resistant mutants and be relatively free of side effects. New nucleosides are in development to try and overcome the thymidine analogue-associated mutations and/or the virus M184V lamivudine mutation so commonly seen in nucleoside-experienced patients. Newer NNRTIs have the potential to be used after efavirenz or nevirapine failure with resistance, but their utility may be related to what mutations are present. Integrase inhibitors have shown great promise but there are issues with resistance. Pharmacokinetic problems especially hepatic enzyme induction or inhibition or simple dosing schedules need to be addressed by new agents. Our understanding of the complex HIV life cycle has led to drugs being developed in new drug classes such as maturation inhibitors or viral interference, Table 1.
Table 1
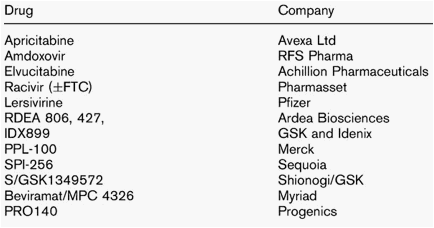
New nucleosides
The reason for developing new nucleoside analogues is not only to improve formulations and to have a better tolerability and toxicity profile but also to be active against common nucleoside resistance mutations. The M184V mutation, seen after virological failure of lamivudine-containing regimens, and the thymidine analogue-associated mutations (or TAMS), usually seen after zidovudine or stavudine, use limit future nucleoside options. Most resistance tests or treatment histories in highly treatment-experienced patients preclude the use of most NRTis in salvage regimens.
Various nucleoside analogues that are in the pipeline give the promise of safety combined with efficacy against common nucleoside-resistant strains.
Apricitabine, Avexa Limited
Apricitabine (ATC; previously known as AVX754, SPD754 or BCH-10618) is a nucleoside reverse transcriptase inhibitor (NRTI) whose mechanism of action is via inhibition of HIV-1 reverse transcription by DNA chain termination.
ATC has activity in vitro against clinical isolates resistant to other NRTIs, including isolates with the M184V mutation, up to five TAMs, as well as L74V, and K65R a mutation seen after virological failure with tenofovir.
Resistance to ATC is slow to emerge in vitro and the resultant mutants have only a low level of resistance to ATC. No resistance to ATC has been seen thus far in treatment-naïve patients during 10 days' monotherapy or in treatment-experienced patients with up to 48 weeks of ATC exposure in combination with other antiretroviral therapy. It appears to have a good safety profile.
It has no significant inhibitory effect upon cytochrome P450 enzymes. P-glycoprotein transport or the glucuronidating enzyme UGT1A1 in vitro.
A Phase 2 dose-ranging clinical study (AVX-201) examined the activity and safety of ATC versus lamivudine in treatment-experienced HIV-1 infected patients with the M184V mutation in reverse transcriptase. ATC was dosed at 600 mg twice daily (b.i.d.) or 800 mg b.i.d. and lamivudine at 150 mg b.i.d. in a 21-day functional monotherapy period, in which both the 600 mg and 800 mg b.i.d. doses of ATC showed better reductions in viral load compared with 150 mg 3TC at this primary endpoint.
Optimization of background antiretroviral therapy occurred at day 21, and but by week 24 the differences between the three groups were small with mean reductions in viral load from day 0 of 2.14, 2.13 and 1.88 log10 copies/ml for the 600 mg ATC, 800 mg ATC and 150 mg 3TC groups, respectively. Both doses of ATC were very well tolerated and showed similar profiles to 3TC. At week 48 the mean reduction in viral load was greater for patients with up to two TAMs at baseline than those with more than two TAMs. Overall, patients with more than two TAMs who received 800 mg ATC appeared to have an improved response compared with those who received 600 mg ATC [1]. Interestingly, in terms of resistance there appears to be a paradox, as most of those with viraemia on ATC maintain the M184V mutation and yet ATC also has activity against this mutant.
A phase 2b/3, randomized, double-blinded dose-confirming study of 800 mg versus 1200 mg b.i.d. versus lamivudine in treatment-experienced patients with the M184V/I mutation is currently recruiting.
DAPD (amdoxovir) RFS Pharma
Amdoxovir (DAPD) is a purine nucleoside analogue. The prodrug is converted to DXG-TP, which is the active metabolite, has activity in vitro against clinical virus isolates containing TAMs at positions 41, 67, 70, 215 and 219 of reverse transcriptase and with the M184V mutation. It does have decreased activity when the K65R mutation is present [2]. It is also active against HIV-2 and hepatitis B virus.
Viral load falls of up to 2 log10 are seen when 500 mg is combined with 200 or 300 mg of AZT taken twice daily for 10 days in patients with or without antiretroviral experience. Viral loads drops were only about half as much if DAPD was taken alone. There were no significant side effects in this short study. Resistance mutations observed at study entry did not change, and no new mutations emerged during the trial [3]. The strategy of combining a new nucleoside with zidovudine because of its side effect profile may only be acceptable when treatment options are severely limited.
Investigators have used DAPD with mycophenolate a selective and reversible inhibitor of de-novo synthesis of deoxyguanosine triphosphate (dGTP). Its effects are selective for lymphocytes and suppress HIV replication through guanine depletion. There were some data to show it increases the efficacy of several reverse transcriptase inhibitors in vitro and in vivo and so 40 adult patients who were highly antiretroviral therapy experienced were given DAPD 500 mg twice daily with placebo or mycophenolate 500 mg twice daily added to their failing antiviral therapy [4].
Median viral load reduction at week 2 was -0.26 log10 copies/ml (P < 0.0001). Response to DAPD/placebo (median -0.37 log10 copies/ml) was greater than to DAPD/mycophenolate (median -0.23 log10 copies/ml), whereas this difference was not statistically significant (P = 0.59). It may have been seen as mycophenolate lowered concentrations of DAPD and its metabolite dioxolane guanosine. Few adverse events were related to study medications, but this strategy does not appear to be a useful for this drug.
Elvucitabine (ACH-126,443, Fd4C) Achillion Pharmaceuticals
In preclinical studies, elvucitabine a L-cytosine analogue was shown to be approximately four-fold more potent in vitro than lamivudine against wild-type HIV and has good activity against strains resistant to other NRTIs and is effective against hepatitis B. Elvucitabine has a long half life of about 100 h and so is dosed at 10 mg once daily. In a 7-day Phase II monotherapy trial in 24 patients, it showed a maximal viral load decrease of 1.73 log10 copies/ml.
ACH-015 was a prospective, randomized, blinded trial in antiviral naïve patients lasting 96 weeks and interim 48-week data were recently presented [5].
Seventy-seven patients were randomized to receive elvucitabine 10 mg daily or lamivudine, 300 mg daily in combination with efavirenz and tenofovir. A total of 55 patients completed 48 weeks of the study and, using ITT analysis, data showed that 65% of patients receiving elvucitabine had a viral load below 50 copies/ml, compared with 78% of those on 3TC more elvucitabine than 3TC patients discontinued, especially earlier on so that by week 12, nine patients had discontinued in the elvucitabine arm and three in the 3TC arm and by week 48 the numbers were 14 and eight respectively. Most the discontinuations, due to adverse events, were not judged to be drug related. The differences may, in part, be related to high lost to follow up rates.
Most salvage regimens contain ritonavir boosted protease inhibitors and recent pharmacokinetic data suggest that, when elvucitabine was coadministered with a single dose of ritonavir, the ritonavir significantly affects the pharmacokinetic of elvucitabine. This finding is probably due to ritonavir decreasing elvucitabine bioavailability by inhibiting influx gut transporters [6].
However, in contrast, when given regularly with ritonavir and lopinavir, although changes in bioavailability after day were variable after day 1 there was an increase in elvucitabine bioavailability. The increase in elvucitabine bioavailability may be due to ritonavir inhibiting in this scenario an efflux gut transporter [7].
To be useful in salvage this drug has to show good activity against nucleoside-resistant strains and data are needed from studies of this compound in highly experienced patients.
Racivir Pharmasset, Inc.
Racivir is an investigational oral, once-daily cytidine nucleoside analogue active against HIV and hepatitis B in laboratory studies. In a Phase I/II study, racivir showed anti HIV activity that lasted more than 2 weeks after the drug was stopped.
In order to assess the benefit of racivir in patients with the M184V mutation, Study 201 was performed. This study was a randomized, double-blinded, placebo-controlled, multicentre trial of 54 patients assessing the safety, tolerability and antiviral effect of a 600 mg dose of racivir. In this study, 42 individuals were randomized to receive either racivir (n = 26) in place of lamivudine or to continue with lamivudine (n = 16) in a double-blinded manner for 28 days. HIV viral loads and genotypes were determined at baseline (mean viral load = 4.1 log) and throughout the study. After the blinded treatment period, individuals were allowed to continue racivir in an open label manner with or without optimized background therapy for an additional 20 weeks, based on their primary care physicians' advice. No serious adverse events attributed to therapy were noted in either group over the initial study period.
At day 28 the mean viral load rose by 0.13 log (a 34.9% increase) in the lamivudine group and dropped by 0.4 log (60.2% reduction) in the racivir group (P = 0.02) [8].
In 14 patients of the racivir-treated group with a M184V mutation and less than three TAMS the mean drop in viral load was greatest in the second week of treatment at 0.7 log, with and 64% of these patients achieving at least a 0.5 log (68%) drop.
Many patients have genotypes that might be responsive to racivir, future studies are being designed to explore this strategy.
Investigational nonnucleoside reverse transcriptase inhibitors
Novel NNRTIs need to be free of toxicities, such as rash, hepatitis and CNS disturbances, have a high genetic barrier and can work against resistance mutations commonly seen after efavirenz and nevirapine. Etravirine has come close to these objectives and others are in development.
Lersivirine (UK-453.061) Pfizer
Lersivirine has been studied in Phase I trials and appears to be active against clinically significant NNRTI mutations: for example K103N, Y181C/I & G190A/S resistance to the drug requires 2-3 mutations.
There is no CYP3A4 inhibition and so drug interactions may not be an important issue, as with other NNRTIs.
It is well tolerated in single doses upto 1800 mg and multiple dose studies up to 1000 mg b.i.d. for 12 days. The maximum tolerated dose has not been identified. The most frequent adverse events seen in these studies were: gastrointestinal and headache and appears generally well tolerated and with no significant laboratory parameters.
A Phase IIa study in 48 HIV-positive patients of 7 days of monotherapy showed that viral load was reduced by up to -1.7 log at the highest doses used 750 mg once daily or 500 mg twice daily [9] There was, no evidence of central nervous system side-effects or rash in this 7-day study.
Further evaluation of UK-453.061 for the treatment of HIV infection is ongoing.
RDEA806 Ardea Biosciences
RDEA806 is a new nonnucleoside reverse transcriptase inhibitor (NNRTI). The medication has been shown to be effective against strains that are resistant to efavirenz, especially those with the K103N mutation, and has an added beneficial effect of reducing uric acid levels. In-vitro resistance mutations selected in the presence of RDEA806 [2] shows reduced phenotypic susceptibility to viruses with five mutations in RT (K104E-E138K-T240I-V179D-F227L) [10].
In a Phase 2 study patients with no preexisting NNRTI or protease inhibitor resistance mutations. Were randomized to four four-treatment arms taken as monotherapy for 7 days and receiving RDEA806 as capsules taken either 400 mg twice daily or 600 mg once daily on an empty stomach or as an enteric-coated tablet formulation either 800 mg once daily with food or 1000 mg once daily on an empty stomach. By day 8, HIV viral load declined by about 1.8 log10 copies/ml across all RDEA809 dose arms, compared with only 0.2 log10 copies/ml in the placebo arm. There was no overall significant dose-response relationship and viral load decline was similar in patients receiving once-daily and twice-daily dosing [11].
No serious adverse events or clinically significant laboratory abnormalities were observed. In particular, rates of rash and CNS symptoms were similar in the RDEA806 and placebo arms.
The major metabolite of RDEA806 is RDEA594, which has no antiviral activity but has a lowering effect on serum uric acid levels. Consequently, RDEA806 is going into a Phase 2a proof-of-concept study for gout.
It is not known if this drug will continue to be developed for HIV as the company state on their website 'we anticipate that the timing of future studies of RDEA806 will be determined in part by the results of our partnering efforts'.
Another NNRTI RDEA427 is from a chemical class that is distinct from RDEA806. On the basis of early preclinical data, RDEA427 may also have even greater activity against a wide range of drug-resistant viral isolates than RDEA806.
IDX899 GSK and idenix pharmaceuticals
IDX899 is a potent nonnucleoside reverse transcriptase inhibitor (NNRTI) being developed for the treatment of HIV-1. It retains marked activity against a broad range of HIV-1 strains, including NNRTI-resistant mutants with single mutations (K103N or Y181C) and double mutations, as well as clinical isolates with documented efavirenz resistance-conferring mutations [12]. Single doses up to 1200 mg of IDX899 and multiple doses of 400 mg b.i.d. and 800 mg daily (q.d.) for 7 days were well tolerated by healthy individuals and no relationship between adverse events and dose has been observed. Clinical laboratory assessments showed no signs of liver or kidney toxicity. The dose of 800 mg daily was then selected as the first dose for a 7-day proof-of-concept study with HIV-1-infected treatment-naïve patients This Phase II proof-of-concept study in 32 HIV-1 infected treatment-naive patients received 800 mg once-daily orally of IDX899 achieved mean viral load reductions of 1.8 log10, after 7 days of treatment [13]. In this study, no treatment-related serious adverse events were reported and no patients discontinued. The most common adverse events observed were dyspepsia, headache and nausea; the rates were similar in those receiving placebo. No significant laboratory abnormalities were observed during the treatment period.
In a press statement in February 2009 GSK stated 'the preliminary clinical evidence of IDX899...warrants continued clinical study as part of GSK's growing drug pipeline in HIV'.
As GSK and Pfizer are involved in a joint HIV venture it will be interesting to know if they will continue to develop this compound and/or lersivirine.
Protease inhibitors in development
Several firms were trying to develop a new type of protease inhibitor that will not be cross resistant with existing drugs. Other goals are to have a favourable metabolic profile and not to require boosting by ritonavir and to have a low pill burden with once daily dosing. Given the large number of existing drugs in this class, relatively few novel protease inhibitors are being developed.
PPL-100 Merck & Co., Inc. (from Jules: my understanding is Merck has discontinued development of this drug)
A Phase 1a single dose pharmacokinetic study in healthy volunteers of PPL-100 showed that the drug may have a half-life of up to 36 h at doses of 2400 mg a day, which may allow for once-daily dosing. Drug exposure with and without ritonavir boosting is currently being evaluated in a phase IIB dose-ranging study, as the drug is a cytochrome p450 3A4 inhibitor. This study is testing PPL-100 doses of 600 and 1200 mg once daily in order to determine whether the drug can be given with a lower pill burden, and without ritonavir, once daily.
In-vitro studies suggest a high genetic barrier to drug resistance as selection of resistance mutations over 52 weeks is limited with appearance of mutations at K45R, M46I, T80I, and P81S.
There appears to be no cross-resistance to amprenavir, lopinavir, atazanavir, saquinavir, indinavir, and nelfinavir [14,15]. Its pharmacokinetic properties may see it used instead of ritonavir as a booster for these drugs as well as a drug in its own right.
SPI-256 Sequoia Pharmaceuticals Inc.
SPI-256 is a novel protease inhibitor that retains low nanomolar activity against most HIV drug-resistant isolates was better than or comparable with darunavir. And has a high genetic barrier to resistance. Passage experiments lead to the L10F, M46I, and I50V mutations occurring [16,17].
A Phase 1 randomized, double-blinded, placebo-controlled, single-dose and escalating-dose crossover study evaluated the safety, tolerability and pharmacokinetics of single, ascending doses of SPI-256 administered alone or in combination with 100 mg ritonavir (RTV) in 59 healthy volunteers.
Peak exposure to SPI-256 given alone was dose proportional over the range of 150-1200 mg and, a single dose of SPI-256 achieved a plasma concentration to suggest the potential of once-daily or twice-daily dosing. When combined with ritonavir, SPI-256 levels were markedly elevated. SPI-256 was generally well tolerated in this healthy volunteer population.
Integrase inhibitors
S/GSK1349572 Shionogi-GlaxoSmithKline pharmaceuticals
S/GSK1349572 is a potent inhibitor of HIV integrase and has limited cross-resistance to raltegravir and elvitegravir-resistant viruses [18·]. In-vitro studies show it was active against HIV from patients experiencing treatment failure with raltegravir. In a multicentre controlled Phase 2a clinical trial, 35 patients not currently receiving antiretroviral therapy were randomly assigned to receive S/GSK1349572 at one of three doses (2 mg, 10 mg, or 50 mg), or placebo after 10 days of treatment, the average HIV viral load fell between 1.51 and 2.46 log10 copies/ml across the three treatment arms compared with a 0.5 log10 copies/ml increase in the placebo arm. The 50 mg group showed nearly 2.5 log10 copies/ml decrease in viral load [19]. No serious adverse events were reported and no patients withdrew from the trial due to side effects or for any other reason. S/GSK1349572 was generally well tolerated and demonstrated unprecedented anti-HIV activity following short-term, once-daily, unboosted dosing in HIV-naïve patients.
CCR5 inhibitors
Several CCR5 inhibitors are in development, but to have a drug that can be used once a day or less frequently and to have as neutral a drug interaction profile as possible are qualities that any new investigational agent would need.
PRO140 progenics pharmaceuticals
PRO140 binds to the extracellular domain of CCR5, in contrast to the small molecule CCR5 antagonists including maraviroc that bind to a hydrophobic pocket formed by the transmembrane helices of CCR5. These small molecule CCR5 antagonists inhibit by allosteric means, that is inhibiting CCR5 by binding away from the active site rather than competitive mechanisms.
This mechanism has some advantages. PRO140 may be active against viruses that are resistant to small molecule CCR5 antagonists. The unique binding pattern of PRO140 means that it inhibits HIV entry without preventing chemokine signalling and, therefore, does not block the natural activity of CCR5. Finally, the difference in binding between the two classes of drug may allow them to work together synergistically.
Originally given intravenously [20], it has also been given subcutaneously in R5 HIV 1-infected individuals in a Phase 2A study [21]. Forty-four HIV-infected individuals were randomized to one of four groups, including a placebo arm, and doses ranging from 162 to 324 mg were given weekly PRO140 subcutaneously weekly on days 1, 8, and 15; after 58 days of follow up, subcutaneous PRO140 demonstrated potent and prolonged antiretroviral activity without significant local or systemic toxicity. These findings are encouraging and demonstrate the potential for infrequent self-administered subcutaneous dosing with PRO140 as an approach to long-acting antiretroviral therapy.
Maturation inhibitors
Beviramat//MPC 4326 Myriad Pharmaceuticals
MPC 4326 also known as beviramat is a maturation inhibitor that targets the Gag capsid SP-1 cleavage site.
Changes at positions 369, 370 or 371 in Gag lead to reductions -0.16 (n = 9), -0.24 (n = 22) and -0.32 logs (n = 11) respectively, compared to -1.08 logs in patients with no changes. It is active in treatment-experienced patient treated with doses ranging upto 400 mg o.d.
Fifty-nine treatment-experienced patients who had a failing (viral load >2000 copies/ml) background regimen received 2 weeks of beviramat or placebo as functional monotherapy [22]. Of 44 patients given the assigned beviramat dose, the mean viral load change was -0.6 log copies/ml vs. +0.05 log for placebo (n = 13 Beviramat has few adverse events the most common being diarrhea (22%; 39%), nausea (20%; 31%) and headache (20%; 23%); all were Grade 1. Gag polymorphisms are common in patients who have received past therapy with multiple anti-HIV treatment regimens. In this study patients who did not have any polymorphisms and so got maximum activity from the drug had a mean fall in their viral load of 1.26 log10.
Phase 3 studies are planned and the analysis of several databases showed that approximately 50% of these patients could benefit from the drug [23].
Other maturation inhibitors are in development.
Other drugs and novel strategies
The cyclophilin inhibitor Debio 025 has activity against both HCV and HIV in preclinical studies, although its anti-HIV effect was not strong in early clinical trials [24].
Gene therapies
The first randomized, double-blinded, placebo-controlled, Phase 2 cell-delivered gene transfer clinical trial has recently been performed [25·].
Patients' haematopoietic progenitor CD34+ stem cells were isolated. and further modified with placebo or a gene coding for tat-vpr-specific anti-HIV ribozyme (OZ1). The enzyme prevents the replication of the HIV virus by excising the tat gene encoded by the viral genome.
Although there was no significant statistical difference between the viral load of OZI and placebo groups at week 48 the time-weighted areas under the curve for the time period 40-48 and 40-100 weeks were found to be substantially less in the OZ1 group. For the entire follow-up period of 100 weeks, the OZ1 group showed an increase in CD4+ lymphocyte count when compared with the placebo group. Those patients who received the OZI cells did not report any treatment-related adverse effect.
This proof-of-concept study is an important development in the development of an effective gene therapy-based strategy for HIV.
Conclusion
Evidence from clinical trials has demonstrated that combining active drugs into a potent regimen based on resistance testing and drug history can lead to suppression of viral load in the majority of so called 'salvage' patients. This strategy has come about because of the availability of drugs in existing classes with potency against common primary mutations and the development of new classes of drugs Patients with few or no active therapy choices should be evaluated frequently as new drugs becoming available to see if suppressive regimens can be constructed. Clinical trials have a pivotal role to play in these patients.
1 Cox S, Southby J. Apricitabine: a novel nucleoside reverse transcriptase inhibitor for the treatment of HIV infection that is refractory to existing drugs. Expert Opin Investig Drugs 2009; 18:199-209.
2 Mewshaw JP, Myrick FT, Wakefield DA, et al. Dioxolane guanosine, the active form of the prodrug diaminopurine dioxolane, is a potent inhibitor of drug-resistant HIV-1 isolates from patients for whom standard nucleoside therapy fails. J Acquire Immune Defic Syndr 2002; 29:11-20.
3 Murphy R, Zala C, Ochoa C, et al. Pharmacokinetics and potent anti-HIV-1 activity of amdoxovir plus zidovudine in a randomized double-blind placebo-controlled study. 15th Conference on Retroviruses and Opportunistic Infections. 3-6 February 2008. Boston. Abstract 794.
4 Margolis DM, Mukherjee AL, Fletcher CV, et al. The use of beta-D-2,6-diaminopurine dioxolane with or without mycophenolate mofetil in drug-resistant HIV infection. AIDS 2007; 21:2025-2032.
5 De Jesus E, et al. Elvucitabine phase II 48 week interim results show safety and efficacy profiles similar to lamivudine in treatment naïve HIV-1 infected patients with a unique pharmacokinetic profile. 48th Interscience Conference on Antimicrobial Agents and Chemotherapy, abstract H-892, Washington, 2008.
6 Colucci P, Pottage JC, Robison H, et al. Effect of a single dose of ritonavir on the pharmacokinetic behavior of elvucitabine, a nucleoside reverse transcriptase inhibitor, administered in healthy volunteers. Antimicrob Agents Chemother 2009; 53:646-650.
7 Colucci P, Pottage JC, Robison H, et al. Multiple-dose pharmacokinetic behavior of elvucitabine, a nucleoside reverse transcriptase inhibitor, administered over 21 days with lopinavir-ritonavir in human immunodeficiency virus type 1-infected subjects. Antimicrob Agents Chemother 2009; 53:662-669.
8 Cahn P, Sosa N, Wiznia A, et al. (racivir 201 Study Team). Racivir demonstrates safety and efficacy in patients harboring HIV with the M184V mutation and <3 TAM. 14th Conference on Retroviruses and Opportunistic Infections; February 25-28, 2007; Los Angeles, California. Abstract 488.
9 Fatkenheuer G, Staszweski S, Plettenberg A, et al. Short-term monotherapy with UK-453,061, a novel NNRTI, reduces viral load in HIV infected patients. Presented at: The International AIDS Conference on HIV Pathogenesis, Treatment and Prevention; 22-25 July 2007; Sydney.
10 Xu W, Zhang Z, Bellows D, et al. Resistance to RDEA806 requires multiple mutations which have limited cross-resistance to other NNRTIs. 48th International Conference on Antimicrobial Agents and Chemotherapy (ICAAC 2008). Washington, DC. 25-28 October 2008. Abstract H-1222.
11 Moyle G, Boffito M, Manhard K, et al. Antiviral activity of RDEA806, a novel HIV nonnucleoside reverse transcriptase inhibitor in treatment naive HIV patients. XVII International AIDS Conference (AIDS 2008). Mexico City. 3-8 August 2008. Abstract THAB0403.
12 Zhou XJ, Pietropaolo K, Damphousse D, et al. Single-dose escalation and multiple-dose safety, tolerability, and pharmacokinetics of IDX899, a candidate human immunodeficiency virus type 1 nonnucleoside reverse transcriptase inhibitor, in healthy subjects. Antimicrob Agents Chemother 2009; 53:1739-1746.
13 Zala C, Murphy R, Zhou X-J, et al. IDX899, a novel HIV-1 NNRTI with high barrier to resistance, provides suppression of HIV viral load in treatment-naive HIV-1-infected subjects. XVII International AIDS Conference (AIDS 2008). Mexico City. 3-8 August 2008. Abstract THAB0402.
14 Wu JJ, Stranix, BR, Milot G, et al. PL-100, a next generation protease inhibitor against drug resistant HIV: in vitro and in vivo metabolism. 44th Interscience Conference on Antimicrobial Agents and Chemotherapy, San Francisco, abstract H-253, 2006.
15 Wu JJ, Dandache1 S, Stranix BR, et al. The HIV-1 protease inhibitor PL-100 has a high genetic barrier and selects a novel pattern of mutations. XV International HIV Drug Resistance Workshop: Basic Principles and Clinical Implications, Sitges, Spain, June 2006.
16 Gulnik S, Afonina E, Eissenstat M, et al. SPI-256, a highly potent HIV protease inhibitor with broad activity against MDR Strains. 13th CROI. Denver USA 2006 Abstract 501.
17 Afonina E, Gulnik S, Yokoe H, Erikson J. In Vitro Selection Strategy and Characterization of Resistance to a Novel Highly potent HIV Protease Inhibitor SPI-256 XV HIV Drug Resistance Workshop, 13-17 June 2006 Sitges, Spain.
18· Underwood M, Johns B, Sato A, et al. S/GSK1349572: a next generation integrase inhibitor with activity against integrase inhibitor resistant clinical isolates from patients experiencing virologic failure while on raltegravir therapy. Fifth IAS Conference on HIV Treatment, Pathogenesis and Prevention, abstract WePeA098, 2009. Appears to be a once-a-day compound that would work against raltegravir-resistant strains.
19 Lalezari J, Sloan L, Dejesus E, et al. Potent antiviral activity of S/GSK1349572, a next generation integrase inhibitor (INI), in INI-naïve HIV-1-infected patients. 5th International AIDS Society Conference on HIV Pathogenesis, Treatment and Prevention (IAS 2009). 19-22 July 2009. Cape Town, South Africa. Abstract TUAB105.
20 Jacobson JM, Thompson MA, Fischl MA, et al. Antiviral activity and tolerability of 5 mg/kg and 10 mg/kg doses of PRO 140, a humanised monoclonal antibody to CCR5. 48th ICAAC, 25-28 October 2008. Washington. Abstract H-1269a.
21 Thompson M, Lalezari J, Saag M, Jacobson J, Zingman B, Stambler N, D'Ambrosio P, Maddon P, Olson W, Morris S. Weekly and Biweekly Subcutaneous PRO 140 Demonstrates Potent, Sustained Antiviral Activity. 16th Conference on Retroviruses and Opportunistic Infections. Montreal, February B-11 2009Abstract 571a.
22 Lalezari J, McCallister S, Gigliotti M, et al. A Phase 2 safety and efficacy study of bevirimat (BVM) in heavily treatment experienced HIV+ patients identifies the target Phase 3 study profile. 48th ICAAC, 25-28 October 2008. Washington. Abstract H-891.
23 McCallister S Lalezari J, Richmond G, et al. HIV-1 gag polymorphisms determine treatment response to beviramat (PA-457). Antivir Ther 2008; 13(Suppl. 3):A10 (abstract no. 8)
24 Klumpp K, Su G, Deval J, et al. 2'-deoxy-nucleoside analogs as potent dual inhibitors of HCV and HIV replication, with selectivity against other viral and human polymerases. 44th Annual Meeting of the European Association for the Study of the Liver (EASL 2009). Copenhagen, Denmark. 22-26 April 2009.
25· Mitsuyasu RT, Merigan TC, Carr A, et al. Phase 2 gene therapy trial of an anti-HIV ribozyme in autologous CD34+ cells. Nat Med 2009; 15:285-292.
|
|
| |
| |
|
|
|