| |
Hepatitis C virus infection: A "liaison a trois" amongst the virus, the host, and chronic low-level inflammation for human survival
|
| |
| |
Download the pdf here
Articles in Press
Jnl of Hepatology
Vincenzo Barnaba
Departimento of Medicina Interna, Sapienza Universita di Roma, Fondazione Andrea Cesalpino, Fondazione Cenci Bolognetti, Italy
Received 28 February 2010; received in revised form 20 May 2010; accepted 9 June 2010. published online 02 July 2010.
Uncorrected Proof
ABSTRACT: Herein, various ambiguous aspects of the immune system that render this complex biological network so highly flexible and able to defend the host from persisting infections such as that induced by the hepatitis C virus (HCV) are reviewed. This ambiguity stems mainly from the property of the immune system to be both protective and harmful. Immunity cannot be fully protective without producing a certain degree of damage (acute hepatitis resulting in resolving HCV infection). In addition, the balance between protection and tissue damage is critical for the development of chronic HCV infection. The establishment of a state of chronic low-level inflammation is instrumental to limit liver immunopathology, to limit viral spread, and ultimately to ensure a long-lasting survival of the host. It is dictated by a fine equilibrium maintained by multiple immunologic mechanisms, including: sensory perception of innate immunity, virus-specific T and B cell functions, control of immune responses, and finally the balance between immunity and immunopathology that has principally evolved to favor the species survival.
Abbreviations: HCV, hepatitis C virus, pDCs, plasmocytoid dendritic cells, cDCs, conventional dendritic cells, PRRs, pattern-recognition receptors, TLRs, toll-like receptors, NOD, intracellular nuclear oligomerisation domain, PAMPs, pathogen-associated molecular patterns, LPS, lipopolysaccharide, IL, interleukin, TRIF, toll-IL-1 receptor domain-containing adaptor inducing IFN-ß, IFN, interferon, DAMPs, damage-associated molecular patterns, UTR, untraslated region, RIG-I, retinoid acid-inducible gene I, IPS-1, adapter molecule IFN-ß promoter stimulator protein 1, p, plasmocytoid, JAK, Janus kinases, E, envelope, NS, non-structural, c, conventional, CCR7, Cys-Cys chemokine receptor 7, TGF, transforming growth factor, Th, T helper, NK, natural killer, KIR, NK cell inhibitory receptor, PD-1, programmed death-1 receptor, TCR, T cell receptor, L, ligand, Treg, T regulatory, Foxp, forkhead box P, IPEX, immunodysregulation polyendocrinopathy enteropathy X-linked, SHPs, Src homology 2-containing tyrosine phosphatases, pSTAT-5, STAT-5 phosphorylation
Conclusions
Through the different (non-mutually exclusive) mechanisms illustrated above, the host survives for a long time in parallel with both the persistent HCV infection and a low-grade liver inflammation that can degenerate into liver failure after several decades. The ambiguous co-existence of virus and inflammation results in an advantage for the evolutionary process and hence for human species survival. If immune responses were invariantly strong and aggressive during a persistent infection such as HCV, they would be unable to eliminate that infection, because of its acquired capacity to escape or to subvert them. In such a situation, exuberant (but non-protective) responses would produce prompt irreversible tissue (hepatic failure) damage, leading to catastrophic epidemic infections. Considering this point of view, chronic (low-level) inflammatory diseases seem to represent a sort of safeguard for the human survival. We can assume that chronic inflammation may be defined as the "Yin and Yang" of the immune system. On the one hand, it guarantees the long-term survival of human hosts despite pathogen persistence. On the other hand, the imbalance of the homeostatic mechanisms maintaining chronic inflammation may degenerate into severe "side-effects" (i.e., the development of either autoimmune diseases or tumours) in a minority of infected individuals. From an evolutionary point of view, the onset of autoimmune diseases or the development of some tumours might be the price to pay following the establishment of chronic inflammation. Indeed, a status of pre-existing chronic inflammation can contribute to the development of cancer, by the production of growth and angiogenic factors eventually promoting cancer-cell survival, implantation, and growth. In addition, chronic inflammation can affect the immune-surveillance directly via its own intrinsic mechanisms (i.e., expansion of Treg cells, T cell exhaustion, etc.), and indirectly by the incapacity to limit the immunosuppressive effects of tumours. The production of soluble factors (i.e., pro-inflammatory or cell growth cytokines) that favor cell proliferation, generally needed for the immune system to defend the host efficaciously, can also facilitate the mitotic cycle of non-lymphoid cells. In the long run, this prolonged stimulation can induce, as in the case of liver cirrhosis by both HBV and HCV, necrosis, cell renewal, and even neoplastic transformation [84]. A further example in HCV infection, is the chronic stimulation of B lymphocytes that can induce the monoclonal expansion of anti-IgG antibodies, which are responsible for the formation of cryoglobulins, autoantibodies, or even the establishment of follicular B cell lymphomas [85].
The immune system simultaneously expresses different strategies that are seemingly opposite but eventually result in an evolutionary advantage. On the one hand, the immune response contributes to species survival; on the other hand it can lead to the sacrifice of single individuals. During the evolutionary process, selective pressure has led to the generation of multiple ambiguous mechanisms to help counteract aggressive infectious agents. Although this is obtained at the cost of severe side-effects (tumour development, autoimmune diseases) in some individuals, these side-effects are considered irrelevant in terms of the survival of the species.
The challenge for scientists is to eliminate the side-effects that emerge in the chronic HCV-host relationship (i.e., cirrhosis, liver failure, HCC, autoimmunity, etc.), possibly via ad hoc modeling and production of new antiviral drugs, immuno-modulatory molecules, therapeutic antiviral antibodies, antiviral small interference (si)RNAs, systems restoring T cell exhaustion (by inhibiting PD-1, Treg cell function, IL-10, or TGF-ß, etc.), and new vaccination strategies.
Article Outline

Introduction
Hepatitis C virus (HCV) is a positive-stranded RNA virus belonging to the Flaviviridae family (reviewed in [1]). HCV eludes host defenses in a considerable portion of infected individuals, developing a status of viral persistence, representing the major cause of chronic hepatitis, cirrhosis, and hepatocellular carcinoma (reviewed in [1], [2], [3]). This review considers the various mechanisms of HCV persistence, and mainly concentrates on those by which T cell responses have been evolved to favor long-term host survival, in spite of chronic HCV-dependent liver disease.
Innate immunity and HCV infection
Resolution of acute infections is dependent on a complex interplay between innate and adaptive immunity. Innate immune cells and molecules play a central role in promptly controlling infections in the early phases and providing the environment required for priming efficient adaptive immune responses. Innate immune cells (mainly monocytes, neutrophiles, dendritic cells [DCs]) are promptly activated upon the recognition of infecting agents by a wide array of pattern-recognition receptors (PRRs), such as the toll-like receptors (TLRs), or the intracellular nuclear oligomerisation domain (NOD)-like receptors [4], [5], [6], [7], [8]. TLRs identify infectious signals derived by molecular patterns common to different pathogens (pathogen-associated molecular patterns [PAMPs], such as lipopolysaccharide [LPS], bacterial DNA, or viral RNA). Then, via their adaptor molecules (i.e., MyD88 for TLR2, 3, 4, 5, 7, 8, 9, 11, and toll-interleukin [IL]-1 receptor domain-containing adaptor inducing interferon [IFN]-ß [TRIF] for TLR3 and 4), they trigger a cascade of down-stream molecules leading to NF-κB and AP-1 activation that ultimately induces the transcription of genes promoting the activities of innate immune cells (cytokine production, maturation, differentiation, migration, etc.). The intracellular NOD-like PRRs recognize dangerous compounds (damage-associated molecular patterns [DAMPs]), such as exogenous crystals (e.g., asbestos causing mesothelioma or asbestosis, silica dust causing silicosis, etc.), or endogenous DAMPs including proteins associated with stressed or dying cells (e.g., uric acid, nucleic acids and their degradation products, such as high-mobility group box 1 protein, oligonucleotides and nucloesides) [7], [9]. It is reasonable hence to postulate that DAMPs derived from hepatocyte necrosis may play a pivotal role in the HCV-dependent liver inflammation.
Importantly, these ancestral signals are also involved in alarming all non-lymphoid nucleated cells (including hepatocytes) that express a more limited repertoire of PRRs than immune cells. Indeed, they quickly respond to infections via the IFN-ß production that provides both an antiviral effect to the infected cells themselves and limits infection of neighboring non-infected cells.
Interferences of HCV with endogenous type I IFN by infected cells
HCV is a single-stranded (ss)RNA virus and, therefore induces type I IFN production in infected cells (i.e., hepatocytes) either upon contact with TLR3 in the endosomal compartments, or upon recognition of the polyuridine motif of HCV 3' untranslated region (UTR) by the retinoid acid-inducible gene I (RIG-I) in the cytoplasm (reviewed in [1]). These processes may be affected by HCV. In vitro studies demonstrated that endogenous HCV-NS3/4A protein cleaves both TRIF and IFN-ß promoter stimulator protein 1 (IPS-1) (adaptor molecules of TLR3 and RIG-I, respectively), thus blocking the down-stream pathway leading to IFN-ß production in transfected hepatocyte cultures (reviewed in [1]). HCV-core protein directly inhibits the down-stream IFN regulatory factor 3 molecule, which in concert with NF-κB, activates IFN-ß gene transcription (reviewed in [1]). This data has been emphasized by observations in vivo revealing that liver biopsies from HCV patients express an inactive form of IPS-1, consistent with it being cleaved [10]. However, patients with acute or chronic HCV infection show normal levels of circulating IFN-α/ß, leading the hypothesis that the latter are not produced by HCV-infected hepatocytes, but by non-infected (likely plasmocytoid [p]DCs) cells. Several HCV proteins interfere with the antiviral signals provided by the cell-surface type I IFN receptors upon engagement by circulating IFNs. The overexpression of HCV-core protein in cell culture interferes with Janus kinases (JAK)/Signal Transducers and Activators of Transcription (STAT) pathway, HCV-envelope (E)2 or non-structural (NS)5A with the function of protein kinase R. HCV-NS5A inhibits 2'-5' oligoadenylate synthetase and induces IL-8 which inhibits induction of the IFN-stimulated genes (reviewed in [1]). In synthesis, HCV seems both to affect the capacity to produce type I IFNs by infected cells and to make the latter less sensitive to the antiviral effect of the same cytokines via disturbing the signals provided by type I IFN receptors (Fig. 1). In vivo models of HCV infection are required to ascertain the importance of these selective defects in providing profound impairment of innate responses in infected cells and ultimately in restraining the priming of adequate adaptive immune responses. Should these mechanisms be demonstrated in vivo, they may take part in the establishment of viral persistence. They may also be amplified by the absence of the genetic polymorphism near the IL28B gene encoding IFN-lambda-3, recently related to both the successful treatment of genotype 1 HCV with IFN-α [11], and the spontaneous resolution in the natural course of HCV infection [12].
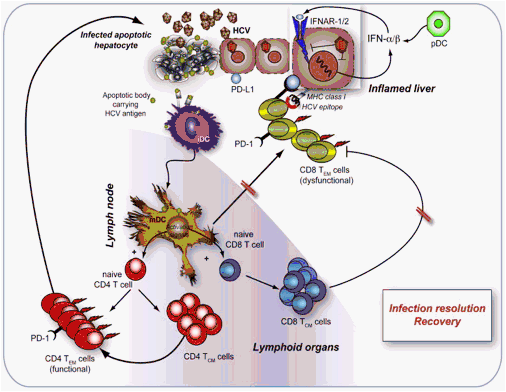
Fig. 1. Immune activities in resolving HCV infection. Innate immunity can be principally affected by HCV at the level of both type I IFN production by infected hepatocytes and the signals provided by the relative receptors (IFNAR-1/2) once they are engaged by soluble type I IFNs (mainly produced by pDCs). If these defects are combined with low viral load or infection by HCV strains that are highly susceptible to antiviral IFN effects, HCV viral spread would be contained, and the functions by DCs, NK, B, and T cells should not be heavily affected. This possibility might account for the evidence that the HCV-specific CD4 T cells efficiently differentiate into protective TEM (with Th1 phenotype) and TCM cells, despite the fact that HCV-specific CD8 TEM cells that result are dysfunctional. Since DCs are not susceptible to HCV infection, they could activate CD8 T cells through the phenomenon of cross-presentation of apoptotic hepatocyte bodies containing HCV products. This phenomenon might not be enough to induce efficient primary or secondary CD8 T cell responses, in the absence of direct HCV presentation by infected DCs. In addition, HCV-specific PD-1+ CD8 T cells (simultaneously recognizing MHC class I/viral epitope complexes and PD-L1 on infected hepatocytes) should acquire an exhausted/dysfunctional phenotype in the site of infection, more than PD-1+ CD4 cells. CD4 T cells might guarantee resolution, by producing protective cytokines, helping antigen-specific B cells, and finally sparing some virus-specific CD8 cells from becoming dysfunctional. Under these conditions, the negative loop leading to T cell exhaustion by the interaction between PD-1 expressed on activated T cells and PD-L1 expressed on all lymphoid and non-lymphoid cells would have the ability to switch off unwanted responses, once the virus has been cleared.
HCV interference in the functions of innate immune cells
Plasmocytoid and conventional dendritic cells
pDCs deriving from the lymphoid lineage represent the most important source of type I IFNs (reviewed in [3]). They produce IFN-α/ß upon engagement of TLR7 and nine by ssRNA and dsRNA, respectively, making them critical players in fighting viruses, particularly in the early phases of infection. However, how HCV can induce IFN-α/ß production by pDCs is unclear [3]. Indeed, TLR7 and nine harbor the endosomal compartments and pDCs (as well as conventional [c]DCs) do not seem permissive to HCV infection, likely because they express CD81 but not claudin-1 that are simultaneously required to allow HCV entry into hepatocytes [13], [14]. Moreover, DC infection by HCV has not been shown by using highly sensitive infection systems, such as recombinant engineer reporter HCV [3]. Another debated question is if pDCs are functionally competent in HCV infection. Despite the contrasting evidence that has been reported on this topic (reviewed in [3]), recent studies that measured functions per pDC basis and not within total PBMCs, revealed no defect in response to TLR stimulation by circulating pDCs of chronically-infected individuals [15], [16]. The functional defects of circulating pDCs, which have been observed upon contact with different non-infecting HCV products in vitro (reviewed in [3]), are difficult to reconcile with the fact that chronically-infected individuals do not display a generalized immuno-dysfunction (they normally respond to other viruses or recall antigens!) and have high levels of endogenous type I IFNs (reviewed in [3]). Therefore, we favor the hypothesis that HCV does not interfere with the pDC functions, but it makes infected hepatocytes non-susceptible to the high levels of circulating pDC-derived type I IFNs, because of its capacity to affect the signals triggered by their own specific type I IFN receptors (Fig. 1).
The second fundamental DC population in humans is constituted by cDCs deriving from the myeloid lineage [17], [18]. Given the critical role of cDCs in priming T cell responses (see Box 1), they have been extensively studied in HCV infection, with the idea that HCV-mediated inhibition of cDC functions could result in inefficient antiviral T cell responses. Contrasting evidence resulted from these analyses. cDCs from chronically-infected individuals have not been found to be numerically decreased in the peripheral blood or even dysfunctional in vitro in terms of pro-inflammatory or antigen-presentation capacities, in all studies or patients [19], [20]. As well as in the case of pDC studies, the relevance of both the analysis on circulating cDCs from patients, and those showing the capability of some recombinant HCV proteins to affect the functions of normal cDCs in vitro [21], [22], is strongly restrained by the evidence that chronically-infected individuals are not globally immuno-compromised. In vivo models of HCV infection are required to determine the possibility of a selective impairment of DCs or HCV-specific T cells infiltrating HCV-infected livers due to the high concentrations of viral proteins produced in the site of infection. The selective dysfunction of liver-infiltrating DCs or T cells might result relevant by profoundly affecting the adaptive immune responses against HCV at the level of the infection site (Fig. 2).

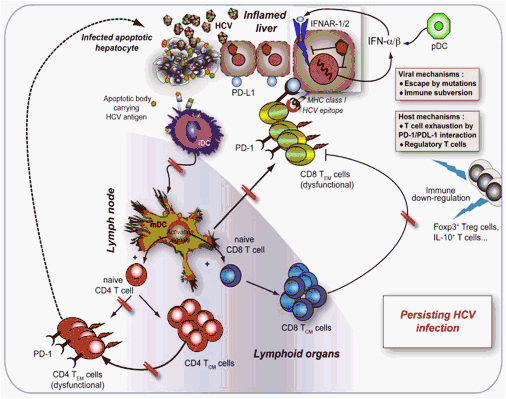
Fig. 2. Immune dysfunctions related to chronic HCV infection. High viral load and/or infection by HCV strains that are not susceptible to antiviral effects of endogenous type I IFNs, facilitating HCV viral spread, might strongly affect the function of DCs, NK, B, and T cells. The misfunction of CD4 and CD8 T cells will result in inefficient effector and memory responses and will result in the development of a state of viral persistence. This would also be conditioned by the emergence of several, non-mutually exclusive factors, such as viral epitope escape, viral subversion, the host immunological mechanisms (PD-1, Treg cells, etc.) addressed to control immunopathology, at the cost of the acquiring side-effects that limit protection. Under these conditions, a state of chronic low-level inflammation will take place and will be instrumental in limiting liver immunopathology, viral spread, and finally to ensure long-lasting survival of the host.
Natural killer (NK) cells
Genetic studies demonstrated the association of some HLA and NK cell inhibitory receptor (KIR) genes with resolution of HCV infection [23]. Functional and molecular studies on HLA-KIR interactions are required to determine if these genetic associations result in blocking particular KIRs expressed by NK cells and hence in NK cell-mediated protection in animal models of HCV infection. New studies have also shown an increased proportion of NK cells expressing activating receptors, enhanced cytotoxic function, and defective cytokine production in chronic HCV infection [24]. Additional investigations should be brought forth to verify if they participate in the establishment of chronic inflammation, on the one hand, and viral persistence, on the other hand [24]. In regards to the reports showing induction of NK cell defects upon exposure to some HCV proteins in vitro [25], [26], in vivo models are needed to ascertain if they effectively play a major role in chronic HCV disease development.
Adaptive immunity and HCV infection
As described above, prompt and efficient innate immune responses are mandatory to prime naïve T or B lymphocytes that will then fight, eliminate, and permanently remember the pathogens encountered, via the specific recognition of microbial epitopes. Successful effector responses and memory establishment by CD4 Th cells are dependent on the presence during priming of a wide array of stimulatory signals: those provided by professional APCs (e.g., DCs) in primis, duration of antigenic stimulus, the cytokine milieu, etc. Priming of protective (cytotoxic) CD8 T cell responses requires the same conditions, but the long-lasting CD8 T cell memory seems to be conditioned by the constant presence of memory CD4 T cells [27]. These mechanisms guarantee the prompt emergence of high frequencies of competent effector T cells that are essential for recovery. Upon infection resolution, effector cells disappear, whereas memory cells remain numerically constant because of the expression of receptors specific for the homeostatic (IL-7 and IL-15) cytokines [28]. The homeostatic proliferation of memory cells in the absence of antigen, is critical for prompt differentiation into effector cells, should they re-encounter the original infecting pathogen.
The immunologic scenario promoting infection resolution is only partially respected in acute HCV infection. However, the common conviction that HCV induces chronic infection in the majority of infected individuals has been challenged by the observation that T cells against multiple HCV epitopes persist in a considerable proportion of healthy (non-infected) individuals accidentally exposed to HCV [29], [30]. This data strongly suggests that recovery from asymptomatic form of HCV infection, and that the generation of efficient virus-specific T cell responses clearing HCV are far more frequent than commonly believed [31].
HCV-specific B and T cell responses
The finding that agammaglobulinemic patients can resolve acute HCV infection upon IFN-α treatment, leads to the hypothesis that HCV-specific T cells may compensate for the lack of neutralizing antibodies to obtain HCV clearance [32]. However, recent data suggested that the prompt emergence of neutralizing antibodies in the early phases of infection could play a major role in clearing HCV in immunocompetent patients. Indeed, they have been detected at high levels both during the early phase of infection in association with spontaneous resolution of HCV [32], and once chronic HCV infection is established (reviewed in [1]). Therefore, the availability of neutralizing antibodies or of appropriate vaccines eliciting them may have a central role in the prophylaxis of HCV infection. It is possible that the role of antibodies may have been underestimated in the past due to methodical difficulties in neutralization assays before the HCVpp system was developed.
The emergence of HCV-specific T cells can be detectable in the peripheral blood or in the liver compartment several weeks after infection in humans or experimental chimpanzee models (reviewed in [1], [3]), corresponding with the initial peak of transaminases and irrespective of clinical outcome (resolution vs. chronicity). Despite the delayed appearance of antigen-specific responses, the latter are essential for the HCV control (reviewed in [1]). The majority of studies have been addressed to analyze CD8 T cells in HCV infection, because of their pivotal role in clearing intracellular pathogens. These studies revealed that the magnitude of CD8 T cell responses does not correlate with the clinical or viral outcome in acute HCV infection [34], [35], [36] (Fig. 1, Fig. 2). HCV-specific CD8 T cells are at a relatively high frequency, but express a dysfunctional phenotype (weak proliferation, IFN-γ production, and cytotoxicity) and increased levels of programmed death-1 receptor (PD-1), known to be associated with the exhausted phenotype, irrespective of infection progression [37], [38], [39], [40], [41], [42]. In contrast to HCV-specific CD8, vigorous responses of HCV-specific CD4 T cells producing IFN-γ and IL-2 (Th1 cell profile) are detectable in the peripheral blood at the time of peak of ALT levels, in patients with acute HCV infection undergoing resolution [35], [36], [43], [44] (Fig. 1). The protective effects of CD4 T cells seem to be due, not only to the antiviral cytokines produced, but also to their capacity to help antiviral B cells and to maintain CD8 T cell memory. Indeed, work in experimental animal models support the idea that the CD4-dependent memory HCV-specific CD8 T cells are indispensable both for HCV control and for providing long-term protection [45], [46]. On the contrary, weak, absent, or transient CD4 responses are correlated with chronic infection progression [35], [36], [43], [44] (Fig. 2), suggesting hence that the simultaneous dysfunctions of both CD8 and CD4 cells are associated with disease progression in the majority of infected individuals. Thus, the combination of functional HCV-specific CD4 and CD8 T cells obviously should be the right recipe for recovery, as it is in resolving flu, CMV, or EBV infections. This scenario may contribute to HCV clearance in a considerable proportion of asymptomatic infected individuals, which have been exposed to a different source of HCV infection [29], [30], [31]. Another aspect to consider is the possibility that other T cell subsets or functions may intervene in dictating the fate of HCV infection. In this context, the role of HCV-specific T (CD4 and/or CD8) cells with a Th17 profile in HCV protection, chronic evolution, or pathogenesis is an important topic requiring more in-depth investigations [47]. Indeed, it has been recently reported that Th17 cells play a key role in establishing chronic viral infections [48], [49]. In the following section, the possible mechanisms that may affect the HCV-specific adaptive immune response will be analyzed.
Mechanisms affecting adaptive immune cells in acute HCV infection
The mechanism of cross-presentation
It would be relevant to determine if the selective impairment of HCV-specific CD8 T cell responses may be related to the observation that DCs are not susceptible to HCV infection, and thus in principle, they cannot process endogenous HCV antigens and directly present the resulting epitopes on class I molecules [3], [13], [14]. As a result, HCV-specific CD8 T cells might be primed only via the mechanism of cross-presentation (see Box 1). In cross-presentation, non-infected DCs capture exogenous HCV antigens or apoptotic liver cells carrying HCV and then cross-present the related HCV epitopes on class I molecules [50], [51], [52], [53] (Fig. 1). This mechanism might not be enough to prime efficient CD8 T cell responses (See Box 2).

PD-1/PD-L1 interaction
A special emphasis has been recently placed on PD-1, a death receptor expressed by T or B cells in the late phases of activation [37], [38]. PD-1 induces peripheral T or B cell tolerance or turns off unwanted immune responses, upon the simultaneous interaction of the T cell receptor (TCR) or BCR with antigens and of PD-1 with its own ligands (L): PD-L1, which is virtually expressed on all somatic cells (particularly from inflamed tissues) and PD-L2, and is mainly expressed by DCs [37], [54]. Inflamed/infected hepatocytes up-regulate expression of both PD-L1 [55] and class I molecules bearing viral epitopes (whereas the class II are undetectable or only barely expressed) [56]. Consequently, HCV-specific effector PD-1+ CD8 (recognizing class I/epitope complexes on infected hepatocytes) more than PD-1+ CD4 cells should acquire an exhausted/dysfunctional phenotype in the site of infection. This may account for the conserved functional capacities of HCV-specific CD4 T cells shown in patients undergoing infection resolution, despite dysfunctional HCV-specific CD8 T cells (Fig. 1). Otherwise, the lack of functionally competent HCV-specific CD4, associated with exhausted CD8 T cells, would unavoidably lead towards chronic infection in the majority of patients (Fig. 2). The yet unresolved question is what makes the total HCV-specific adaptive (both CD4 and CD8) T cell responses "not-functionally-competent". This is likely conditioned by the emergence of several, non-mutually exclusive factors, such as high viral load, viral epitope escape, viral subversion, and host immunological mechanisms (PD-1, T regulatory [Treg] cells, etc.) addressed to control immunopathology, at the cost of the side effect of limiting protection. All these factors will induce the generation of non-protective virus-specific CD4 and CD8 T cells, which might even become harmful (Fig. 2).
Viral mutations
HCV's strong capability to mutate B or T cell epitopes and possibly to escape related responses at several levels (antigen processing, MHC binding, TCR or BCR recognition, etc.) is due to its high replication rate and the lack of proofreading capacity of its polymerase (reviewed in [1]). The evidence that about 50% of the CD8 epitopes continue to escape [57], [58] leaving another 50% that do not mutate, renders the role of mutational escape in HCV persistence unclear. Despite both the huge T or B cell repertoire and the fact that several viral epitopes do not mutate due to fitness constraints (reviewed in [1]), there are some CD8 escape mutations associated with fitness costs [59], [60], [61]. These, in synergy with additional mechanisms (high viral load, viral subversion, host immune-suppressive mechanisms, etc.), may participate in the establishment of viral persistence, particularly during the course of the acute phase of infection when the highest level of selective pressure occurs (reviewed in [1]).
Virus-induced immune-subversion
As mentioned above, the studies revealing a general subversion of both T cell and DC functions, upon exposure to some HCV proteins in vitro [21], [22], [62], [63], are difficult to reconcile with the fact that chronically-infected individuals are not globally immuno-compromised. However, if in vivo models of HCV infection demonstrate a selective impairment of T cells infiltrating HCV-infected livers due to the high concentrations of viral proteins produced at the site of infection, this may participate in establishing HCV persistence by affecting the local adaptive immune responses.
Regulatory cytokines
A recent report demonstrated that peripheral HCV-specific CD4 and CD8 T cells producing IL-10 are detectable in the early phases of acute HCV infection [64]. These cells seem to suppress antiviral effector responses, promoting chronic evolution of infection, while limiting progressive liver damage [64]. These suggestions are reminiscent of our previous studies showing that IL-10 producing HCV-specific CD8 T cells infiltrate the liver of chronically-infected individuals [65]. They inversely correlated with both the frequency of HCV-specific T cells producing IFN-γ and the inflammatory staging at the level of liver biopsies, suggesting that they modulate excessive liver immunopathology [65]. Accordingly, it has been reported that intrahepatic HCV-specific IL-10 producing CD8 T cells prevent liver damage during chronic infection [66]. TGF-ß-producing virus-specific CD4 and CD8 T cells have been related to antiviral immune suppression and chronic HCV infection evolution [67]. Taken together, these data suggest that regulatory cytokines such as IL-10 or TGF-ß minimize liver inflammation, at the cost of the protective immune responses clearing the virus (Fig. 2).
CD25+Foxp3+ Treg cells
Treg cells expressing the transcription factor forkhead box P (Foxp)3 develop either in the thymus (natural) or in the periphery from conventional CD4+ T cells (induced) [68], [69], [70], [71]. A lack of Foxp3 expression results in the complete absence of Treg cells, which leads to the development of severe autoimmunity, as observed in immunodysregulation polyendocrinopathy enteropathy X-linked (IPEX) syndrome [68], [69], [70], [71]. The main physiological functions of Treg cells are as follows: (a) to participate in the establishment of peripheral tolerance by inhibiting autoreactive T or B lymphocytes that escaped either thymus or bone marrow checkpoints, respectively (central tolerance), (b) to suppress ongoing protective immune responses once they are no longer necessary or become harmful after the elimination of the pathogen and (c) to limit excessive immunopathology during chronic inflammatory diseases. As a result of the expression of the Il-2 gene-inhibitory Foxp3 transcription factor, Treg cells do not produce IL-2 and are unable to respond to antigens (anergy) [72], [73]. However, Foxp3 activity maintains high levels of IL-2 receptors (CD25hi) on Treg cells, hence compensating for the incapacity of producing IL-2. Indisputably, CD25hi Treg cells promptly proliferate both in vitro and in vivo in response to relevant antigens in the presence of paracrine IL-2, which is mainly produced by responder (effector) T-lymphocytes, but it is dominantly absorbed by Treg cells expressing higher CD25 levels than responder T cells [74], [75], [76]. This appears to represent a key suppression mechanism, because CD25hi Treg cells steal the majority of IL-2 produced by responder T cells that in turn will be deprived of their most important growth factor. Furthermore, Treg cells suppress via different, likely non-mutually exclusive mechanisms, involving membrane molecules (such as cytotoxic T-lymphocyte antigen-4 or adenosine receptors) and suppressive cytokine production (such as TGF-ß or IL-10) [68], [69], [70], [71]. Treg cells are induced and proliferate in response to HCV and seem to modulate liver inflammation in the course of chronic infection [77], [78]. Therefore, the model of HCV infection supports the idea that Treg cells participate in the establishment of a fine equilibrium between immunopathology and immune protection, ultimately resulting in the long-lasting survival of the host during chronic infections [69], [70], [79], [80], [81], [82], [83] (Fig. 2). This would be dependent on a compromise between a status of chronic low-level hepatic inflammation and the generation of antiviral responses that, although unable to clear HCV, are enough to limit excessive viral spread. It is unclear how Treg cells control unwarranted inflammation without completely suppressing the protective immune responses. High CD25 expression by Treg cells drives a positive feedback loop, as the dominant IL-2 capture increases STAT-5 phosphorylation (pSTAT-5) that in turn drives Treg cell proliferation and function. We recently showed that PD-1 is over-expressed on Foxp3+ Treg cells and limits Treg cell proliferation and function during chronic HCV infection. The expression of PD-1, upon contact with its own ligands, inhibits pSTAT-5 via the activation of Src homology 2-containing tyrosine phosphatases (SHPs) [78] (Fig. 3). As a consequence, responder T cells can escape from excessive expansion of Treg cells and render them available for responding to possible novel waves of infection. This negative feedback loop assumes a different significance during chronic infections, such as HCV. The incapacity to clear HCV by the immune system (due to the various mechanisms emphasized above) perpetuates a vicious spiral, whereby responder T cells are chronically stimulated to produce IL-2 that will be dominantly adsorbed by CD25hi Treg cells that in turn will continuously suppress the effector responses. PD-1 up-regulation limits the excessive expansion of Treg cells by controlling pSTAT-5 and fine-tunes Treg function in order to minimize the immunopathology without completely switching off those intended to limit excessive viral spread (Fig. 3). This may represent a critical contra-suppression mechanism that has evolved to assure that Treg cells have limited suppression. Homeostatic balance participates in establishing a status of chronic low-level liver inflammation that is in turn instrumental to ensure long-lasting survival of the host.
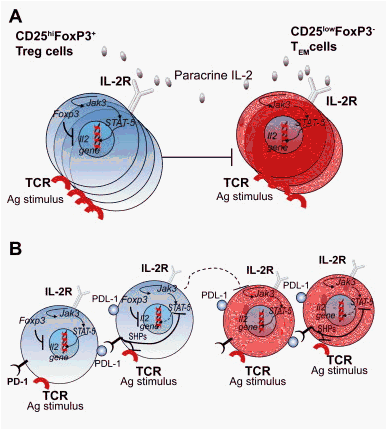
Fig. 3. PD-1 controls Treg cells in HCV infection. (A) Responder (CD25lowFoxp3-) TEM cells proliferate in response to HCV antigens, and produce IL-2, which through IL-2R (CD25) signaling, induces pSTAT-5. This leads to the development of the genetic program dictating their effector phenotype. In parallel, the same phenomena occurs for the (CD25hiFoxp3+) Treg cells that do not proliferate to viral antigens alone because of the expression of the Il-2 inhibitory gene Foxp3. Their proliferation is dependent on the dominant capture of paracrine IL-2 that is initially produced by responder T cells. CD25hiFoxp3+ Treg cells can proliferate by the engagement of the Jak3/STAT-5 pathway, and extrinsically down-regulate the TEM cell responses. (B) In the late phases of T cell activation, the death receptors intrinsically deliver negative signals to activated T cells (including Fas, CTLA-4, and PD-1) in order to terminate the T cell responses. PD-1 is up-regulated on both responder and Treg cells and upon contact with PD-L1/2 inhibits pSTAT-5 possibly via SHP2. This mechanism results in limiting both TEM cell responses and excessive Treg cell function. Under conditions resulting in HCV resolution, this loop is self-limited because of the disappearance of the viral antigenic stimuli. During a chronic HCV infection, in which responder T cells have been unable to clear HCV, the negative loop is maintained by the persisting HCV antigens that chronically stimulate IL-2 producing responder T cells. Chronic PD-1 expression on both TEM and Treg cells modulate the potential excessive pSTAT-5-dependent cell proliferation. The resulting contra-regulation of Treg cells will have an important role in limiting excessive suppression of immune responses, controlling the spreading virus at the cost inability to maintain chronic low-level liver immunopathology. This mechanism establishes long-lasting survival of the host.
Conclusions
Through the different (non-mutually exclusive) mechanisms illustrated above, the host survives for a long time in parallel with both the persistent HCV infection and a low-grade liver inflammation that can degenerate into liver failure after several decades. The ambiguous co-existence of virus and inflammation results in an advantage for the evolutionary process and hence for human species survival. If immune responses were invariantly strong and aggressive during a persistent infection such as HCV, they would be unable to eliminate that infection, because of its acquired capacity to escape or to subvert them. In such a situation, exuberant (but non-protective) responses would produce prompt irreversible tissue (hepatic failure) damage, leading to catastrophic epidemic infections. Considering this point of view, chronic (low-level) inflammatory diseases seem to represent a sort of safeguard for the human survival. We can assume that chronic inflammation may be defined as the "Yin and Yang" of the immune system. On the one hand, it guarantees the long-term survival of human hosts despite pathogen persistence. On the other hand, the imbalance of the homeostatic mechanisms maintaining chronic inflammation may degenerate into severe "side-effects" (i.e., the development of either autoimmune diseases or tumours) in a minority of infected individuals. From an evolutionary point of view, the onset of autoimmune diseases or the development of some tumours might be the price to pay following the establishment of chronic inflammation. Indeed, a status of pre-existing chronic inflammation can contribute to the development of cancer, by the production of growth and angiogenic factors eventually promoting cancer-cell survival, implantation, and growth. In addition, chronic inflammation can affect the immune-surveillance directly via its own intrinsic mechanisms (i.e., expansion of Treg cells, T cell exhaustion, etc.), and indirectly by the incapacity to limit the immunosuppressive effects of tumours. The production of soluble factors (i.e., pro-inflammatory or cell growth cytokines) that favor cell proliferation, generally needed for the immune system to defend the host efficaciously, can also facilitate the mitotic cycle of non-lymphoid cells. In the long run, this prolonged stimulation can induce, as in the case of liver cirrhosis by both HBV and HCV, necrosis, cell renewal, and even neoplastic transformation [84]. A further example in HCV infection, is the chronic stimulation of B lymphocytes that can induce the monoclonal expansion of anti-IgG antibodies, which are responsible for the formation of cryoglobulins, autoantibodies, or even the establishment of follicular B cell lymphomas [85].
The immune system simultaneously expresses different strategies that are seemingly opposite but eventually result in an evolutionary advantage. On the one hand, the immune response contributes to species survival; on the other hand it can lead to the sacrifice of single individuals. During the evolutionary process, selective pressure has led to the generation of multiple ambiguous mechanisms to help counteract aggressive infectious agents. Although this is obtained at the cost of severe side-effects (tumour development, autoimmune diseases) in some individuals, these side-effects are considered irrelevant in terms of the survival of the species.
The challenge for scientists is to eliminate the side-effects that emerge in the chronic HCV-host relationship (i.e., cirrhosis, liver failure, HCC, autoimmunity, etc.), possibly via ad hoc modeling and production of new antiviral drugs, immuno-modulatory molecules, therapeutic antiviral antibodies, antiviral small interference (si)RNAs, systems restoring T cell exhaustion (by inhibiting PD-1, Treg cell function, IL-10, or TGF-ß, etc.), and new vaccination strategies.
|
|
| |
| |
|
|
|