| |
Abnormalities of lipid metabolism in hepatitis C virus infection
|
| |
| |
Gut
Published Online First 26 July 2010
doi:10.1136/gut.2009.192732
Clinical impact
The multifaceted interactions between HCV and lipid metabolism may not only have biological significance in the HCV life cycle. The potential consequences on the host can hardly be overlooked, but should not be overemphasised either.
The effect of steatosis on liver fibrosis seems to depend on the pathogenesis of fat accumulation rather than on the presence of steatosis per se. There is good evidence that steatosis is a risk factor of liver disease progression. This has been shown by both cross-sectional71 72 91-93 and longitudinal studies.72 94-96 However, the steatosis observed in genotype 3, presumably viral in origin, does not seem to be independently associated with liver fibrosis.93 In this same meta-analysis, fibrosis was independently associated with steatosis only in patients with genotype 1.93 Thus, the accelerated fibrogenesis may depend on the co-occurrence of steatosis of non-viral origin. In patients with chronic hepatitis C who do not drink alcohol and are infected with non-3a genotypes, the most frequent correlate of fatty liver is increased body weight. Being overweight or presenting with a visceral obesity are two independent risk factors for fatty liver.72 97 Its pathogenesis is most likely the insulin resistance98 via several mechanisms leading to an imbalance between uptake, de novo synthesis and degradation of fatty acids by hepatocytes resulting in excess triglyceride accumulation.54 In addition, if insulin resistance is included in a multivariate logistic regression to analyse the factors independently associated with fibrosis, the association between steatosis and fibrosis disappears in favour of the association with insulin resistance.97 Thus, although HCV may cause massive steatosis, this does not seem to lead to accelerated fibrogenesis and, if any association between fatty liver and fibrosis progression exists, this appears to depend on the co-occurrence of a steatosis caused by factors other than HCV.
Whether HCV-induced steatosis is a risk factor for the appearance of hepatocellular carcinoma (HCC) remains an open question. Some transgenic mice models of HCV-induced fatty liver have shown progression to HCC.82 99 The production of reactive oxygen species may be responsible for somatic mutations in these models.82 100 Human studies are, however, inconclusive. In patients with chronic hepatitis C, steatosis may be an independent risk factor for the development of HCC101, but a retrospective study including a smaller number of patients with chronic hepatitis C did not confirm these findings.102 Thus, further prospective studies are warranted to evaluate the role of HCV-associated steatosis in liver carcinogenesis.
The interaction of HCV with lipid metabolism may have three consequences regarding the treatment of hepatitis C with antiviral agents. First, since HCV interacts with the LDL receptor during uptake by hepatocytes, one may speculate that high levels of circulating LDL might interfere with hepatocyte infection by HCV. Although the competition has been shown in vitro,17 there are no data confirming that this occurs also in the human infection. It is unknown whether this potential receptor competition may account for the repeated observation that elevated LDL levels are associated with an increased response to IFNα.103-106
A second issue is the impact (if any) of virally-induced steatosis on the response to treatment. It has been shown in large clinical trials that steatosis impairs the response to antiviral therapy.74 The effect, however, is more pronounced in patients with non-3a genotype,74 hinting again at insulin resistance as the mechanism affecting the response to IFNα and suggesting that the viral steatosis does not impair response to treatment.107 108 The pretreatment insulin resistance score is indeed a predictor of a poor response to treatment.10 The molecular reasons for the correlation between insulin and IFNα resistance are unclear. It is worth noting that patients with chronic hepatitis C with virally-induced steatosis do not have increased insulin resistance levels relative to patients without steatosis.98
A third and final point worthy of discussion is the interesting potential use of 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors in HCV therapy. It was mentioned above how use of lovastatin may inhibit HCV RNA replication in hepatoma cells.46 In an in vitro assay the anti-HCV activity of several statins were compared.50 Fluvastatin exhibited the strongest activity, atorvastatin and simvastatin an intermediate degree and lovastatin showed the weakest inhibitory activity.50 A few clinical trials have used statins as monotherapy or in combination with the standard of care (SOC). In a pilot trial, atorvastatin was administered as monotherapy (at the conventional daily dose of 20 mg) to 10 patients with chronic hepatitis C with hypercholesterolaemia.109 Although serum cholesterol and LDL decreased, there was no effect on serum HCV RNA after 4 and 12 weeks of treatment.109 Similarly disappointing results were reported in 42 patients coinfected with HCV and HIV and receiving fluvastatin 80 mg daily, in whom the expected decrease of serum cholesterol and LDL was paralleled by a paradoxical increase in HCV RNA levels.110 However, in another study conducted on 31 veterans, different oral doses of fluvastatin (20-320 mg/day) induced a modest viral suppression which was sometimes short-lived but nonetheless significant.111 Statins lower the levels of LDL and enhance LDL receptor expression; whether the increased HCV RNA observed in the study by Milazzo et al110 may depend on the facilitation of HCV hepatocyte uptake remains to be proven. It is noteworthy, however, that use of statins does not reduce the efficacy of SOC, as suggested by two recent encouraging trial results. In another small uncontrolled trial, 20 mg/day fluvastatin was given in addition to the pegylated IFNα/ribavirin combination to a group of 21 patients. In the 15 patients who received a 48-week course of therapy the SVR was 67%.112 All the above studies show that statin use is safe in patients with hepatitis C, but also that the choice of the appropriate statin may be critical in order to achieve antiviral effects. The relationship between concomitant use of statins and the lipid profile, with particular regard to the serum level of LDL and total cholesterol, was evaluated retrospectively in a very large trial.113 The results show that statin use was associated with significantly greater chances of reaching SVR, independent of the baseline lipid profile. In particular, statin users had higher chances of SVR when aged >48 years, of non-African American ethnicity and female. Further research on this issue is justified.
---------------------
Abnormalities of lipid metabolism in hepatitis C virus infection
Gut
Published Online First 26 July 2010
doi:10.1136/gut.2009.192732
Francesco Negro
Divisions of Clinical Pathology and of Gastroenterology and Hepatology, University Hospital, Geneva, Switzerland
Correspondence to Francesco Negro, Divisions of Clinical Pathology and of Gastroenterology and Hepatology, University Hospital, rue Gabrielle-Perret-Gentil 4, 1211 Gen�ve 4, Switzerland; francesco.negro@hcuge.ch
Abstract
Hepatitis C virus (HCV) is a human pathogen responsible for acute and chronic liver disease, infecting an estimated 130-170 million persons worldwide. An intriguing feature of HCV infection is its peculiar relationship with lipids: (1) HCV virions circulate in serum bound to lipoproteins; (2) lipids have been shown to modulate (and, indeed, are essential to) the HCV life cycle; and (3) an occasionally severe accumulation of triglycerides is found in a distinct subgroup of patients in the form of hepatic steatosis. As a result, lipid metabolism is overall altered, conferring an idiosyncratic profile to HCV infection. The scope of this review is to discuss these aspects, focusing on both their molecular mechanisms and their clinical consequences.
Summary box
In hepatitis C virus (HCV) infection, the lipid metabolism is characterised by the following peculiar features:
* HCV virions circulate in serum bound to triglyceride-rich lipoproteins.
* Lipids modulate (and indeed are essential to) the HCV life cycle.
* An occasionally severe hepatocyte steatosis is observed in a subgroup of patients with chronic hepatitis C.
The clinical relevance of these findings is debated:
* Virally-induced steatosis does not seem to accelerate the clinical and histological progression of liver disease or to reduce responsiveness to antiviral drugs.
* Available data on the antiviral properties of cholesterol synthesis inhibitors are not univocal and warrant further studies.
Introduction
Hepatitis C virus (HCV) is a positive strand RNA virus of the Flaviviridae family that infects about 130-170 million people (ie, ∼2.2-3% of the world population).1 Infection with HCV induces both innate and adaptive immune responses that resolve the infection in 15-45% of individuals.2 Failure to clear the virus leads to persistent infection, in most cases associated with chronic progressive liver damage (chronic hepatitis C). Treatment with the combination of pegylated interferon-α (IFNα) and ribavirin results in sustained clearance of HCV in 45-80% of chronically infected individuals.3-5 The morbidity and mortality associated with chronic hepatitis C are mainly attributable to progression towards cirrhosis and hepatocellular carcinoma.6 As a consequence, HCV-induced end-stage liver disease is currently the leading indication for liver transplantation in most western countries (box 1).
Box 1 Major facts about the hepatitis C virus (HCV)
* HCV is a positive strand RNA virus (genus Hepacivirus within the Flaviviridae family).
* is estimated to infect about 130-170 million persons (∼2.2-3% of the world population).
* Primary HCV infection persists in 55-85% of individuals.
* Persistent HCV infection is mostly associated with chronic hepatitis.
* Progression of chronic hepatitis C leads to cirrhosis (5-30% over 2-3 decades) and hepatocellular carcinoma (2-4% yearly in patients with established cirrhosis).
* Treatment of chronic hepatitis C with a combination of pegylated interferon-α and ribavirin results in sustained clearance of HCV in 45-80% of cases.
* HCV-induced end-stage liver disease is the leading indication for liver transplantation in most western countries.
LIPIDS & HCV
The spectrum of severity of chronic hepatitis C varies widely, as does the rate of its progression to the cirrhotic stage. This heterogeneity largely depends on host and environmental factors,7 although the contributing role of viral features such as the HCV genotype has recently been revisited.8 Cofactors influencing hepatitis C severity and progression include age, gender, excess alcohol consumption, coinfections with other hepatotropic viruses and/or HIV and the metabolic syndrome.7 The role of the latter in the pathogenesis of hepatitis C has attracted considerable attention in recent years. The metabolic syndrome has reached pandemic proportions and, based on estimates, it will become a major cause of morbidity and mortality worldwide in the near future.9 The relationship between HCV and the metabolic syndrome is important because of the multifaceted direct molecular interactions between HCV and lipid metabolism.10 11 In particular, HCV infection is characterised by a peculiar relationship with lipids: (1) HCV virions circulate in serum bound to lipoproteins; (2) lipids have been shown to modulate (and, indeed, are essential for) the HCV life cycle; and (3) an occasionally severe accumulation of triglycerides in hepatocytes is observed in a distinct subgroup of patients in the form of fatty liver. In summary, lipid metabolism shows widespread alterations, conferring an idiosyncratic profile to HCV infection. This review will discuss these aspects, focusing on both their molecular mechanisms and their clinical consequences.
HCV and lipoproteins
HCV was found to circulate bound to lipoproteins soon after its discovery. In a landmark study, Thomssen and collaborators reported that HCV virions failed to band homogeneously across a sucrose density gradient, with HCV RNA-positive fractions being distributed over a wide range, extending from a high density (1.20 g/cm3) to an unusually low density (1.03 g/cm3).12 Interestingly, low-density fractions, at variance with high-density ones, could be precipitated by anti-β-lipoprotein antisera, raising the hypothesis that virions could be tightly associated with such lipoproteins.12 In addition, not only could low-density fractions transmit HCV to experimental animal models,13 but the infectious titre of HCV-positive inocula was inversely correlated with the buoyant density of HCV RNA-carrying particles,14 hinting at a critical role for the association with lipoproteins in conferring infectivity. Low-density HCV particles were subsequently also shown to be infectious in vitro15 and in natural human infections.16 Furthermore, cell culture grown HCV successfully transmitted to the chimpanzee is associated with low-density fractions.15
By electron microscopy, these low-density particles are roughly spherical with a heterogeneous diameter of 50-150 nm.17 Chemically, they are highly enriched in triglycerides17 and can be almost completely precipitated by anti-apolipoprotein B (apoB) and anti-lipoprotein E (apoE) antibodies,18 hence the name lipoviroparticles (LVP).17 In addition, apolipoproteins CII and CIII have been reported to be associated with LVP.19 LVP contain HCV RNA and all of the structural viral proteins.17-19 In particular, the viral envelope proteins E1 and E2 are present at the surface of LVP and can thus be recognised by anti-envelope antibodies under non-denaturating conditions.19 The observation that LVP contain both apoB isoforms (ie, apoB-100 and apoB-48)19 is worth noting because, although apoB-100 is synthesised by hepatocytes, apoB-48 expression is specific to enterocytes where it is incorporated in chylomicrons. This raises the issue of the participation of enterocytes in the HCV life cycle. In support of this hypothesis, HCV non-structural antigens have been detected in small intestine epithelial cells in a proportion of viraemic HCV carriers.20 In addition, LVP surge and are rapidly enriched in triglycerides after a fat-rich meal.19 21
The association of HCV with lipoproteins appears to have a precise biological significance. HCV uptake by hepatocytes is mediated, among other proteins,11 by the low-density lipoprotein (LDL) receptor.22 This phenomenon was initially shown in culture where the endocytosis of HCV and other members of the Flaviviridae family correlated with the LDL receptor expression and was competed out both by antibodies against anti-LDL receptor and by other specific inhibitors of the LDL/LDL receptor interaction.22 In keeping with the fact that both apoB-100 and apoE are ligands for the LDL receptor is the important observation that anti-apoB-100 and anti-apoE antibodies inhibit HCV cell binding17 22 and in vitro infection.23 24 The likely heterogeneity of LVP size is not an issue because the long flexible modular structure of the extracellular N-terminal domains of the LDL receptor are designed to allow binding to triglyceride-rich lipoproteins of different sizes (figure 1).
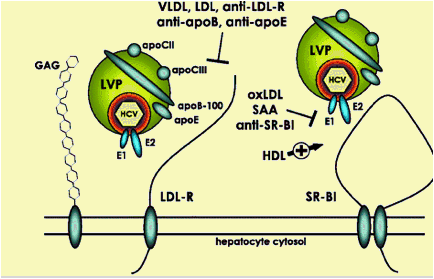
Figure 1
Interaction between the hepatitis C virus (HCV) lipoviroparticles (LVP) and the lipoprotein receptors at the hepatocyte surface. The initial attachment involves glycosaminoglycans (GAG) and the low-density lipoprotein receptor (LDL-R). Binding to abundant unbranched GAG, that is the prosthetic groups of membrane proteoglycans, is low-affinity and non-specific. Conversely, binding to the flexible structure of the extracellular LDL-R domains is mediated by apoB-100 and apoE and can be inhibited by very low-density lipoproteins (VLDL), LDL and by antibodies directed against LDL-R, apoB and apoE. Binding to the scavenger receptor B type I (SR-BI) is facilitated by high-density lipoprotein (HDL) and occurs via the viral envelope protein E2; the SR-BI binding sites for HDL and E2 are, however, distinct. Oxidised LDL (oxLDL), serum amyloid A (SAA) and antibodies against SR-BI inhibit the binding of LVP to SR-BI. Neither the attachment to LDL-R nor the binding to SR-BI are sufficient to establish a productive HCV infection which requires the interaction of HCV particles with several other cell surface proteins, such as the tetraspanin CD81 and the tight junction proteins claudin-1 and occludin (not shown in the figure). For a thorough discussion of HCV receptors, see Popescu and Dubuisson.
HCV cell entry is also mediated by another lipoprotein receptor, the scavenger receptor class B type I (SR-BI),25-28 which is responsible for the entry of various classes of lipoproteins, primarily the high-density lipoproteins (HDL). The interaction is determined by the binding to the viral envelope protein E2.25 However, the SR-BI determinants that bind to HDL are different from those that bind the viral envelope. HDL mediate enhancement of HCV infection via SR-BI, but this does not occur via a direct binding of HDL to HCV particles.29 Although HDL enhance the efficiency of HCV infection, anti-SR-BI antibodies and SR-BI-specific siRNA inhibit HCV infection independently of these lipoproteins.27 Besides providing a docking site for HCV particles, the SR-BI facilitates HCV entry also through its physiological function (ie, cholesterol uptake and/or efflux). Thus, there appears to be some specific SR-BI-mediated lipid transfer function(s) hijacked by HCV to favour infection.29 Contrary to HDL, another SR-BI ligand-oxidised LDL-inhibits HCV entry.30 Finally, serum amyloid A (SAA), an acute phase protein synthesised by the liver, blocks HCV entry by interacting directly with virions,31 and this inhibitory effect is prevented by co-incubation of SAA with HDL,31 suggesting a close interaction between these two proteins in modulating HCV infection. The final internalisation of HCV by target cells, however, occurs only upon interaction of viral particles with further cell surface receptors, and the mere binding of HCV to the LDL receptor and the SR-BI is not sufficient to support productive infection.11
The tight association of circulating HCV to triglyceride-rich lipoproteins may be accounted for by the fact that HCV particles exploit the very low-density lipoprotein (VLDL) assembly and export pathway to be released from hepatocytes (figure 2). Intracellular infectious viral particles are similar in size but differ in their buoyant density (1.15-1.20 g/ml) from secreted particles (1.03-1.16 g/ml), as determined by ultracentrifugation.32 This suggests that infectious HCV particle composition is modified during release from hepatocytes. HCV assembly and maturation occur in the endoplasmic reticulum, and both depend on the microsomal triglyceride transfer protein (MTP) and apoB.33 MTP participates in the VLDL assembly by uploading lipids to the nascent apoB polypeptide after this has been translocated into the endoplasmic reticulum lumen. Inhibitors of MTP activity such as the compound BMS-200150,33 the grapefruit flavonoid naringenin34 or other substances35 significantly decrease the secretion of HCV from infected cells. Similar results can be obtained by inhibiting apoB synthesis with short interfering RNAs.33-35 Thus, it is likely that HCV co-opts VLDL assembly and secretion pathways, although the finer details of this interaction are currently unclear and subject of intense speculation.
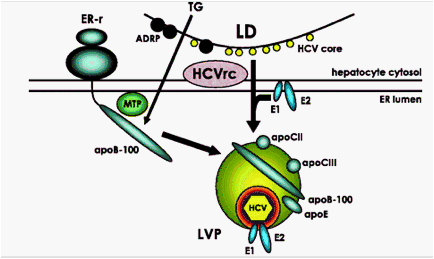
Figure 2
Simplified schematisation of some of the factors participating in hepatitis C virus (HCV) lipoviroparticle (LVP) assembly. Soon after synthesis and processing the HCV core protein is transferred to the lipid monolayer of the lipid droplet (LD) surface, gradually displacing resident proteins such as the adipocyte differentiation-related protein (ADRP) (black circles). The core protein also directs the transfer of the HCV replication complex (HCVrc) to the LD surface where the assembly of virions is supposedly taking place. Meanwhile, endoplasmic reticulum (ER)-associated ribosomes (ER-r) synthesise apoB-100 which is transferred to the ER lumen. Here the microsomal triglyceride transfer protein (MTP) uploads triglycerides (TG) to the apoB to form TG-rich lipoproteins. HCV virions, consisting of core protein, the RNA genome and the two envelope proteins E1 and E2, bud through the ER lipidic bilayer merging with nascent lipoproteins. The intermediate steps of this process are hypothetical and largely unknown. The final LVP, whose size can vary from 50 to 150 nm, contains both the HCV virion proper and the VLDL constituents (see text for details).
The interaction between HCV morphogenesis and cellular lipids is not limited to the final assembly of LVP but starts early on, soon after viral polyprotein synthesis. The core protein is tightly associated with lipid droplets (LD), and this physical interaction is mediated by specific amino acids within the central D2 domain of the viral protein.37 Immediately after its synthesis the core protein is cleaved by a signal peptidase and a signal peptide peptidase, giving rise to a mature form that is transferred from the endoplasmic reticulum membrane to the surface of LD.38 Hydrophobic residues determine the stability of this binding,39 40 a critical event in determining the subsequent efficient virus assembly.40 The binding of HCV core to LD also alters the intracellular distribution of LD which are relocated to the perinuclear area.41 LD redistribution can be prevented by disrupting the microtubule network or by blocking the dynein motor protein, and this results in reduced virion production.41 In addition, the core protein gradually displaces the proteins physiologically found at the surface of the LD such as the adipocyte differentiation-related protein.41 Once at the surface of the LD, the HCV core recruits other viral non-structural proteins.42 43 LD-enriched fractions contain HCV RNA, and virus particles have been observed in close proximity to LD.42 In a series of elegant electron microscopy-based observations, HCV-like particles have been seen budding from convoluted endoplasmic reticulum membrane structures in close proximity to LD.44 The interaction between HCV proteins and the LD therefore appears to be critical for virion assembly, and LD may be the specialised organelles hijacked to provide the triglycerides necessary for the assembly of LVP (figure 2).
Lipids and the HCV life cycle
Lipids are essential to the HCV life cycle: they may exert their effect at different levels-that is, as prosthetic groups of viral proteins and/or of cellular cofactors of HCV replication, as components of the specialised membrane structure where HCV replication takes place or, as discussed above, as constituents of LVP.
The acute HCV infection of chimpanzees is associated with an increased intrahepatic expression of genes involved in lipidogenesis45 such as the ATP citrate lyase, an enzyme activated by the transcription factor sterol-responsive element binding protein (SREBP).45 In the subgenomic HCV replicon system it was shown that 25-hydroxycholesterol, which inhibits SREBP cleavage/translocation, causes a decrease in fatty acid biosynthesis and HCV replication.45 In contrast, nystatin, which may activate the SREBP pathway through cholesterol sequestration, caused a dose-dependent increase in replication levels by nearly 100%.45 HCV RNA replication in hepatoma cells can be disrupted by treatment with lovastatin, a drug that decreases the production of mevalonate by inhibiting 3-hydroxy-3-methylglutaryl-CoA reductase.46 Mevalonate is a precursor of hydrophobic prenyl prosthetic groups such as the geranylgeranyl group which is necessary to anchor various proteins to cell membranes. The cholesterol-biosynthetic pathway controls HCV RNA replication by regulating the cellular levels of geranylgeranyl pyrophosphate, and the impact of geranylgeranylation depends on the fatty acid content of the cell.47 The inhibition of HCV RNA replication by lovastatin can be overcome by the addition of geranylgeraniol, suggesting that HCV RNA replication requires one or more geranylgeranylated proteins.46 47 One such protein was identified as the F-box and leucine-rich repeat-containing protein 2 (FBL2).48 Knockdown of FBL2 mRNA is parallelled by inhibition of HCV replication in vitro.49 Other statins possess anti-HCV activity in vitro,50 and their activity is reversed by mevalonate or geranylgeraniol.50
In addition to cholesterol precursors, fatty acids can also stimulate (or inhibit) HCV replication. Interestingly, their effect seems to depend on their degree of saturation.47 Polyunsaturated fatty acids, including arachidonic acid, docosahexaenoic acid and eicosapentaenoic acid, inhibit HCV replication.51 On the contrary, saturated and monounsaturated fatty acids stimulate it.47 Inhibition of fatty acid synthase (FAS) by cerulenin45 or C7552 blocks HCV replication in a subgenomic replicon system and the secretion of virions in the JFH1 infectious system. Fatty acids can be used as prosthetic groups in exactly the same way as cholesterol intermediates. These prosthetic groups may facilitate viral non-structural protein anchoring to the specialised membrane structures that serve as the platform for HCV RNA replication and assembly. For example, the non-structural protein 4B undergoes a double palmitoylation on the two cysteine residues 257 and 261 at its C-terminal end. Site-directed mutagenesis has shown that these lipid modifications, particularly of Cys261, are important for protein-protein interactions in the formation of the HCV RNA replication complex.53 Thus, palmitoylation may be another post-translational modification essential to the HCV life cycle.
The fatty acids necessary for HCV replication and assembly must be provided in sufficient quantity by the infected cell. Although there are situations where the uptake of fatty acids by hepatocytes may be pathologically increased, such as in the insulin-resistant state,54 there is good experimental evidence that HCV directly stimulates lipidogenesis. During primary infection in chimpanzees, HCV activates genes involved in lipid metabolism via SREBP.45 In vitro, HCV induces transcriptional activation, proteolytic processing and phosphorylation of the two factors SREBP-1c and SREBP-2, responsible for the transactivation of enzymes involved in the synthesis of fatty acids and cholesterol, respectively.55 SREBP activity is stimulated in vitro by several viral proteins including the core55 and the non-structural proteins 256 and 4B.55 57 Activation of SREBP and several enzymes involved in lipidogenesis has also been reported in transgenic mice expressing different HCV proteins.58 59 In addition to activating SREBP, the HCV core protein may also bind to and activate the DNA-binding domain of the retinoid receptor α, a transcriptional regulator that controls many cellular functions including cellular lipid synthesis.60 Enzymes responsible for fatty acid synthesis such as the acetyl-Coa carboxylase 151 and the FAS56 61 are, indeed, strongly activated by the expression of viral proteins in vitro. Unfortunately, the rare data on the expression level of these factors in the liver of chronic hepatitis C patients have suggested an association between the activation of SREBP-1c and the severity of inflammation and fibrosis, but not steatosis.62 Similarly, intrahepatic FAS mRNA levels were not correlated with steatosis,62 albeit that they have been reported to be increased in two recent reports.63 64 This suggests that some caution should be used in interpreting data when these are primarily obtained in experimental models. Metabolic pathways and responses to external stimuli (such as viral infection) may not be comparable to what occurs in human livers, especially when these models involve metabolically inefficient tumour cell lines. Thus, further large-scale studies using appropriate experimental settings and designs are warranted.
HCV and fatty liver
The fact that HCV stimulates lipidogenesis raises the issue of the long-known relationship between hepatitis C and fatty liver. The prevalence of fatty liver in patients with chronic hepatitis C varies between 40% and 80%, depending on the prevalence of alcohol consumption, overweight, type 2 diabetes and other risk factors of fatty liver.65 If all these factors of fatty liver are excluded, the prevalence of steatosis in chronic hepatitis C is still approximately 40%-that is, about a twofold increase compared with chronic hepatitis B.66 67 This suggests that HCV may directly cause fatty liver, at least in a subgroup of patients with chronic hepatitis C. Indeed, the association is so strong that in the pre-serology era it was used as a diagnostic tool to identify patients with chronic non-A, non-B hepatitis.68 69
Steatosis is more frequent and severe in patients with HCV genotype 3,70-72 suggesting the presence of specific sequences across the genome of genotype 3 that are involved in fat accumulation within hepatocytes. This is further supported by two additional observations. First, the severity of steatosis correlates with the level of HCV RNA, both in liver71 and in serum,72 especially in patients with genotype 3. Second, the fatty liver may significantly decrease-if not disappear altogether-when patients are successfully treated with antiviral agents, especially in patients with genotype 3.71 73 74 Steatosis may persist in most patients with non-3 genotypes, even in case of sustained virological response (SVR).74 Interestingly, in anecdotal cases where this has been carefully documented, the recurrence of HCV after the end of therapy may result in the reappearance of steatosis in patients in whom it had disappeared during treatment.75 There are several experimental models of HCV-induced steatosis including in vitro transient expression systems76-79 and transgenic mice.58 80-82 The constructs vary as to the viral proteins expressed and the genotype but, in most cases, the core protein seems sufficient to induce steatosis, the genotype 3a being the most efficient.76-78 Some of these models have been used to identify the mechanisms responsible for the triglyceride accumulation. As discussed above, in several experimental systems HCV enhances lipogenesis via the activation of specific transcription factors. This would be a plausible mechanism, sufficient to explain the fatty liver often seen in patients with chronic hepatitis C. It is disturbing that the rare data available from patients' livers have so far failed to confirm this.61 However, HCV may cause fatty liver via other independent mechanisms, and one of them may be the impaired lipoprotein secretion. It is noteworthy that the serum levels of apoB and cholesterol are reduced in patients with chronic hepatitis C in whom steatosis later responds to antiviral therapy.83 This suggests that, at least in patients with steatosis, HCV may interfere with the VLDL assembly and/or secretion. To further confirm this, the disappearance of fatty liver in SVR upon successful antiviral therapy coincides with the normalisation of cholesterol and ApoB levels.74 84 In a transgenic mouse model, the HCV core protein was found to inhibit MTP activity.81 As discussed above, this enzyme plays a key rate-limiting role in VLDL assembly. Thus, its inhibition would lead to the accumulation of triglycerides otherwise uploaded onto VLDL, and the morphological counterpart of this would be hepatocyte steatosis. Interestingly, the intrahepatic levels of MTP mRNA are reduced in patients with chronic hepatitis C, especially those with steatosis and/or genotype 3,85 thus reinforcing the view that HCV-induced steatosis may be due to an impaired MTP function. There is no satisfactory explanation as to why-at least in some patients-HCV should inhibit an enzymic activity that is so critical to its life cycle, as seen above. Another pathway that may be perturbed by HCV leading to fat accumulation is fatty acid oxidation. Transfection of hepatoma cells with the HCV core protein is followed by a reduced expression of peroxisome proliferator-activated receptor α (PPARα), a nuclear receptor regulating several genes responsible for fatty acids degradation.86 In keeping with these in vitro data, intrahepatic PPARα mRNA is significantly reduced in patients with chronic hepatitis C87 88 and the carnitine palmitoyl acyl-CoA transferase 1A, a target gene of PPARα responsible for mediating the long chain fatty acids transport across the mitochondrial membrane, is downregulated by HCV both in vitro86 and in the liver of patients with chronic hepatitis C.89 Finally, an additional mechanism leading to fatty liver may be an incresaed afflux of non-esterified fatty acids to hepatocytes, as is the case in the insulin resistance syndrome.54 Although HCV may be associated with extrahepatic insulin resistance,90 this seems to involve striated muscles rather than the adipose tissue. Thus, there is no evidence as yet that HCV may induce fatty liver via an increased lipolysis in adipocytes. A schematic summary of the suggested models of HCV-induced fatty liver is presented in figure 3.
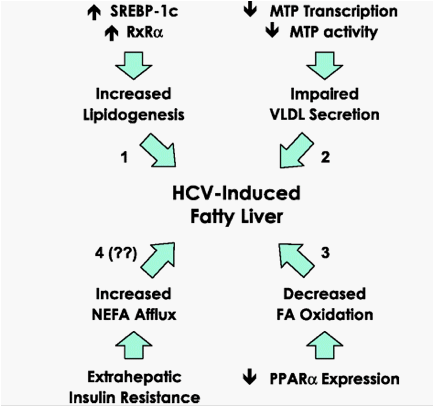
Figure 3
Schematic representation of the proposed mechanisms of hepatitis C virus (HCV)-induced fatty liver. (1) Increased lipogenesis occurs via activation of specific transcription factors, largely documented in experimental models but so far not supported by the scanty findings in human livers. (2) Impaired secretion of very low-density lipid (VLDL) is supported by both experimental data and analysis of human liver tissue: in particular, microsomal triglyceride transfer protein (MTP) mRNA and activity seem reduced in case of steatosis. (3) Decreased fatty acid (FA) oxidation is compatible with the reduced expression of peroxisome proliferator-activated receptor α (PPARα) in the liver of patients with chronic hepatitis C. (4) Although an increased afflux of non-esterified fatty acids (NEFA) may lead to steatosis, there is no evidence that this occurs in patients with hepatitis C without the metabolic syndrome.
The sequence responsible for fatty accumulation is not definitively known. Some data suggest that a phenylalanine at position 164 of the core sequence found in genotype 3a but replaced by a tyrosine in all other genotypes may be associated with activation of FAS76 and accumulation of large lipid droplets in hepatocytes.78 Other microheterogeneities in other HCV genomic regions, or even host factors, may modulate the steatosis phenotype.79
Clinical impact
The multifaceted interactions between HCV and lipid metabolism may not only have biological significance in the HCV life cycle. The potential consequences on the host can hardly be overlooked, but should not be overemphasised either.
The effect of steatosis on liver fibrosis seems to depend on the pathogenesis of fat accumulation rather than on the presence of steatosis per se. There is good evidence that steatosis is a risk factor of liver disease progression. This has been shown by both cross-sectional71 72 91-93 and longitudinal studies.72 94-96 However, the steatosis observed in genotype 3, presumably viral in origin, does not seem to be independently associated with liver fibrosis.93 In this same meta-analysis, fibrosis was independently associated with steatosis only in patients with genotype 1.93 Thus, the accelerated fibrogenesis may depend on the co-occurrence of steatosis of non-viral origin. In patients with chronic hepatitis C who do not drink alcohol and are infected with non-3a genotypes, the most frequent correlate of fatty liver is increased body weight. Being overweight or presenting with a visceral obesity are two independent risk factors for fatty liver.72 97 Its pathogenesis is most likely the insulin resistance98 via several mechanisms leading to an imbalance between uptake, de novo synthesis and degradation of fatty acids by hepatocytes resulting in excess triglyceride accumulation.54 In addition, if insulin resistance is included in a multivariate logistic regression to analyse the factors independently associated with fibrosis, the association between steatosis and fibrosis disappears in favour of the association with insulin resistance.97 Thus, although HCV may cause massive steatosis, this does not seem to lead to accelerated fibrogenesis and, if any association between fatty liver and fibrosis progression exists, this appears to depend on the co-occurrence of a steatosis caused by factors other than HCV.
Whether HCV-induced steatosis is a risk factor for the appearance of hepatocellular carcinoma (HCC) remains an open question. Some transgenic mice models of HCV-induced fatty liver have shown progression to HCC.82 99 The production of reactive oxygen species may be responsible for somatic mutations in these models.82 100 Human studies are, however, inconclusive. In patients with chronic hepatitis C, steatosis may be an independent risk factor for the development of HCC101, but a retrospective study including a smaller number of patients with chronic hepatitis C did not confirm these findings.102 Thus, further prospective studies are warranted to evaluate the role of HCV-associated steatosis in liver carcinogenesis.
The interaction of HCV with lipid metabolism may have three consequences regarding the treatment of hepatitis C with antiviral agents. First, since HCV interacts with the LDL receptor during uptake by hepatocytes, one may speculate that high levels of circulating LDL might interfere with hepatocyte infection by HCV. Although the competition has been shown in vitro,17 there are no data confirming that this occurs also in the human infection. It is unknown whether this potential receptor competition may account for the repeated observation that elevated LDL levels are associated with an increased response to IFNα.103-106
A second issue is the impact (if any) of virally-induced steatosis on the response to treatment. It has been shown in large clinical trials that steatosis impairs the response to antiviral therapy.74 The effect, however, is more pronounced in patients with non-3a genotype,74 hinting again at insulin resistance as the mechanism affecting the response to IFNα and suggesting that the viral steatosis does not impair response to treatment.107 108 The pretreatment insulin resistance score is indeed a predictor of a poor response to treatment.10 The molecular reasons for the correlation between insulin and IFNα resistance are unclear. It is worth noting that patients with chronic hepatitis C with virally-induced steatosis do not have increased insulin resistance levels relative to patients without steatosis.98
A third and final point worthy of discussion is the interesting potential use of 3-hydroxy-3-methylglutaryl-CoA reductase inhibitors in HCV therapy. It was mentioned above how use of lovastatin may inhibit HCV RNA replication in hepatoma cells.46 In an in vitro assay the anti-HCV activity of several statins were compared.50 Fluvastatin exhibited the strongest activity, atorvastatin and simvastatin an intermediate degree and lovastatin showed the weakest inhibitory activity.50 A few clinical trials have used statins as monotherapy or in combination with the standard of care (SOC). In a pilot trial, atorvastatin was administered as monotherapy (at the conventional daily dose of 20 mg) to 10 patients with chronic hepatitis C with hypercholesterolaemia.109 Although serum cholesterol and LDL decreased, there was no effect on serum HCV RNA after 4 and 12 weeks of treatment.109 Similarly disappointing results were reported in 42 patients coinfected with HCV and HIV and receiving fluvastatin 80 mg daily, in whom the expected decrease of serum cholesterol and LDL was paralleled by a paradoxical increase in HCV RNA levels.110 However, in another study conducted on 31 veterans, different oral doses of fluvastatin (20-320 mg/day) induced a modest viral suppression which was sometimes short-lived but nonetheless significant.111 Statins lower the levels of LDL and enhance LDL receptor expression; whether the increased HCV RNA observed in the study by Milazzo et al110 may depend on the facilitation of HCV hepatocyte uptake remains to be proven. It is noteworthy, however, that use of statins does not reduce the efficacy of SOC, as suggested by two recent encouraging trial results. In another small uncontrolled trial, 20 mg/day fluvastatin was given in addition to the pegylated IFNα/ribavirin combination to a group of 21 patients. In the 15 patients who received a 48-week course of therapy the SVR was 67%.112 All the above studies show that statin use is safe in patients with hepatitis C, but also that the choice of the appropriate statin may be critical in order to achieve antiviral effects. The relationship between concomitant use of statins and the lipid profile, with particular regard to the serum level of LDL and total cholesterol, was evaluated retrospectively in a very large trial.113 The results show that statin use was associated with significantly greater chances of reaching SVR, independent of the baseline lipid profile. In particular, statin users had higher chances of SVR when aged >48 years, of non-African American ethnicity and female. Further research on this issue is justified.
Conclusions
When it comes to its relationship with lipid metabolism, HCV is a remarkable virus. Its interaction with lipoproteins and its ability to induce massive steatosis are quite unique and idiosyncratic. HCV is clearly exploiting the host lipid metabolism to its advantage. Whether we may turn this interaction to our advantage remains to be proven. It should be acknowledged that much of what we know about this topic comes from simple but careful observations made in the clinical setting (in some cases even long before HCV was discovered, such as the identification of virally-induced fatty liver). To follow on this path, the preliminary data on the clinical use of inhibitors of the cholesterol synthesis are encouraging and may thus pave the way for further work.
|
|
| |
| |
|
|
|