| |
Raltegravir SWITCHMRK Study Published in The Lancet jan 13 2010
- publication pdf attached
|
| |
| |
Download the PDF here
"The SWITCHMRK studies showed that switching from lopinavir-ritonavir to raltegravir was associated with greater reductions in serum lipid concentrations than was continuation of lopinavir-ritonavir in HIV-infected patients with stable viral suppression on lopinavir-ritonavir-based combination therapy. In the heterogeneous population enrolled in the studies, a higher rate of HIV suppression was achieved in patients who continued lopinavir-ritonavir than in patients who switched to raltegravir, especially in patients who had virological failure before entry.....By contrast, patients without previous virological failure had similar viral suppression rates in both treatment groups......
.....Our finding that substitution of lopinavir-ritonavir for raltegravir did not achieve non-inferiority compared with continuation of lopinavir-ritonavir underscores the complex considerations involved in providing the best possible treatment regimens for individual patients. To understand the different virological response rates in the treatment groups of the SWITCHMRK trials, we undertook subgroup analyses. The prespecified set of subgroup analyses yielded virological results generally consistent with the overall results. However, when specific features of antiretroviral treatment history were examined from a retrospective collection of supplementary data, a plausible explanation for the virological results favouring lopinavir-ritonavir in the overall study population emerged. Participants whose lopinavir-ritonavir-based regimen at screening was their first regimen or patients without previous virological failure had similar virological response rates at week 24 in both treatment groups. Baseline resistance testing could not be done because patients needed to have undetectable vRNA concentrations at screening; however, these subgroups of patients were likely to have been receiving at least two active reverse transcriptase inhibitors in their background therapy, providing adequate antiretroviral support for raltegravir. Our results are consistent with those of the STARTMRK trial,20 in which treatment-naive patients received two active agents in addition to raltegravir, and the findings of the BENCHMRK trials,18, 19 in which treatment-experienced patients who received two or more active agents in combination with raltegravir had better virological responses than did those who received less active background regimens."
"112 (32%) patients in the raltegravir group and 123 (35%) patients in the lopinavir-ritonavir group had virological failure on a previous antiretroviral regimen. 38 (5%) patients (raltegravir, n=17; lopinavir-ritonavir, n=21) had vRNA concentration 50 copies per mL or more at baseline (median 100·5 copies per mL [IQR 66·0-193·0]) despite screening for concentrations less than 50 copies per mL before randomisation"
Table 2. Proportion of patients with viral RNA concentration less than 50 copies per mL at week 24 in the SWITCHMRK studies
Switch to a raltegravir-based regimen versus continuation of a lopinavir-ritonavir-based regimen in stable HIV-infected patients with suppressed viraemia (SWITCHMRK 1 and 2): two multicentre, double-blind, randomised controlled trials
The Lancet, Early Online Publication, 13 January 2010
Prof Joseph J Eron MD a, Benjamin Young MD b, Prof David A Cooper MD c, Michael Youle MD d, Edwin DeJesus MD e, Jaime Andrade-Villanueva MD f, Cassy Workman MD g, Roberto Zajdenverg MD h, Prof Gerd Fatkenheuer MD i, Daniel S Berger MD j, Princy N Kumar MD k, Anthony J Rodgers MS l, Melissa A Shaughnessy MS l, Monica L Walker BS l, Richard JO Barnard PhD l, Michael D Miller PhD l, Mark J DiNubile MD l, Bach-Yen Nguyen MD l, Randi Leavitt MD l, Xia Xu PhD l, Dr Peter Sklar MD l Corresponding AuthorEmail Address, for the SWITCHMRK 1 and 2 investigators
Summary
Background
To reduce lipid abnormalities and other side-effects associated with antiretroviral regimens containing lopinavir-ritonavir, patients might want to switch one or more components of their regimen. We compared substitution of raltegravir for lopinavir-ritonavir with continuation of lopinavir-ritonavir in HIV-infected patients with stable viral suppression on lopinavir-ritonavir-based combination therapy.
Methods
The SWITCHMRK 1 and 2 studies were multicentre, double-blind, double-dummy, phase 3, randomised controlled trials. HIV-infected patients aged 18 years or older were eligible if they had documented viral RNA (vRNA) concentration below the limit of assay quantification for at least 3 months while on a lopinavir-ritonavir-based regimen. 707 eligible patients were randomly allocated by interactive voice response system in a 1:1 ratio to switch from lopinavir-ritonavir to raltegravir (400 mg twice daily; n=353) or to remain on lopinavir-ritonavir (two 200 mg/50 mg tablets twice daily; n=354), while continuing background therapy consisting of at least two nucleoside or nucleotide reverse transcriptase inhibitors. Primary endpoints were the mean percentage change in serum lipid concentrations from baseline to week 12; the proportion of patients with vRNA concentration less than 50 copies per mL at week 24 (with all treated patients who did not complete the study counted as failures) with a prespecified non-inferiority margin of -12% for each study; and the frequency of adverse events up to 24 weeks. Analyses were done according to protocol. These trials are registered with ClinicalTrials.gov, numbers NCT00443703 and NCT00443729.
Findings
702 patients received at least one dose of study drug and were included in the efficacy and safety analyses for the combined trials (raltegravir, n=350; lopinavir-ritonavir, n=352).
Percentage changes in lipid concentrations from baseline to week 12 were significantly greater (p<0·0001) in the raltegravir group than in the lopinavir-ritonavir group in each study, yielding combined results for total cholesterol -12·6% vs 1·0%, non-HDL cholesterol -15·0% vs 2·6%, and triglycerides -42·2% vs 6·2%.
At week 24, 293 (84·4%, 95% CI 80·2-88·1) of 347 patients in the raltegravir group had vRNA concentration less than 50 copies per mL compared with 319 (90·6%, 87·1-93·5) of 352 patients in the lopinavir-ritonavir group (treatment difference -6·2%, -11·2 to -1·3). Clinical and laboratory adverse events occurred at similar frequencies in the treatment groups. There were no serious drug-related adverse events or deaths. The only drug-related clinical adverse event of moderate to severe intensity reported in 1% or more of either treatment group was diarrhoea, which occurred in ten patients in the lopinavir-ritonavir group (3%) and no patients in the raltegravir group. The studies were terminated at week 24 because of lower than expected virological efficacy in the raltegravir group compared with the lopinavir-ritonavir group.
Interpretation
Although switching to raltegravir was associated with greater reductions in serum lipid concentrations than was continuation of lopinavir-ritonavir, efficacy results did not establish non-inferiority of raltegravir to lopinavir-ritonavir.
Funding
Merck.
Discussion
In SWITCHMRK 1 and 2, HIV-infected patients with stable viral suppression on lopinavir-ritonavir-based combination therapy who switched to raltegravir had greater reductions in concentrations of triglycerides, total cholesterol, and non-HDL cholesterol from baseline to week 12 than did patients who continued on lopinavir-ritonavir. However, the efficacy results of the individual studies did not establish non-inferiority of raltegravir to lopinavir-ritonavir, measured by the proportion of patients with vRNA concentration less than 50 copies per mL at week 24. In the combined analysis of the two studies, switching to raltegravir was associated with a lower virological response rate at week 24 than was continuation of lopinavir-ritonavir, leading to early termination of the trials. Virus resistant to raltegravir was detected in most assessable patients who developed virological failure on raltegravir, and the mutational patterns were consistent with genotypes previously reported in treatment-experienced and treatment-naive patients who had treatment failure with this drug.14, 19, 20, 25 Both study drugs were generally well tolerated, and no deaths or serious drug-related adverse events occurred during the trials.
In large phase 3 studies of raltegravir in treatment-experienced18 and treatment-naive patients,20 raltegravir-based combination regimens rapidly suppressed HIV RNA concentrations below the limit of detection in most patients. In heavily pretreated patients infected with multiclass-resistant virus, virological response rates were better when raltegravir was combined with two or more other active agents; results were less robust but still substantial when raltegravir was combined with a single fully active drug or no active drugs.19 In phase 2 and 3 clinical trials, modest changes in serum concentrations of cholesterol and triglycerides were seen up to at least 48 weeks of raltegravir treatment.15-18,20 In view of the drug's antiretroviral efficacy in treatment-naive and treatment-experienced patients together with minor lipid effects and overall tolerability, a study of raltegravir as a substitute for a ritonavir-boosted protease inhibitor in a suppressive combination regimen was a logical step in the exploration of raltegravir use in patients with HIV infection.
Our finding that substitution of lopinavir-ritonavir for raltegravir did not achieve non-inferiority compared with continuation of lopinavir-ritonavir underscores the complex considerations involved in providing the best possible treatment regimens for individual patients. To understand the different virological response rates in the treatment groups of the SWITCHMRK trials, we undertook subgroup analyses. The prespecified set of subgroup analyses yielded virological results generally consistent with the overall results. However, when specific features of antiretroviral treatment history were examined from a retrospective collection of supplementary data, a plausible explanation for the virological results favouring lopinavir-ritonavir in the overall study population emerged. Participants whose lopinavir-ritonavir-based regimen at screening was their first regimen or patients without previous virological failure had similar virological response rates at week 24 in both treatment groups. Baseline resistance testing could not be done because patients needed to have undetectable vRNA concentrations at screening; however, these subgroups of patients were likely to have been receiving at least two active reverse transcriptase inhibitors in their background therapy, providing adequate antiretroviral support for raltegravir. Our results are consistent with those of the STARTMRK trial,20 in which treatment-naive patients received two active agents in addition to raltegravir, and the findings of the BENCHMRK trials,18, 19 in which treatment-experienced patients who received two or more active agents in combination with raltegravir had better virological responses than did those who received less active background regimens.
Major protease-inhibitor mutations are rarely selected by treatment failure when boosted protease-inhibitor regimens are used as first-line therapy.6-10,26 Lopinavir-ritonavir monotherapy either as initial or as simplification therapy maintains viral suppression in around 60-85% of patients.27-29 As with ritonavir-boosted protease inhibitors in general, lopinavir-ritonavir has a high genetic barrier to resistance. Long-term monotherapy with raltegravir has not been studied; however, in the BENCHMRK studies,19 functional monotherapy sustained suppression in approximately 50% of patients. Since patients who had received previous antiretroviral therapy and patients with a history of virological failure were not excluded from the SWITCHMRK studies, this implied difference in efficacy between lopinavir-ritonavir and raltegravir when used as the only fully active agent could partly explain the 6% between-treatment difference in response rates at week 24 seen in our trials. However, resistance to reverse transcriptase inhibitors was documented at the time of virological failure in only five of eight assessable patients in the raltegravir group who were infected with raltegravir-resistant virus.
Efficacious antiretroviral regimens can be modified for a variety of reasons, such as better tolerability or lower toxicity of the new regimen, as well as convenience or simplification. The SWITCHMRK studies were double-blind trials in which treatment was switched, rather than simplified. The studies have similarities to and differences from earlier studies of switching versus continuation of stable therapy. All participants in the SWITCHMRK trials were changed to a more complicated regimen with an increased number of pills, possibly reducing adherence and affecting responses in both treatment groups. The double-dummy design helped to limit bias and prevent the differential dropout across treatment groups that was seen in the SLOAT trial.30 The SWAN31 and ATAZIP32 studies were randomised, open-label trials in which treatment was switched (within the same drug class) to simpler regimens but eligibility was restricted on the basis of previous virological failure or resistance, possibly accounting for the higher suppression rates in these studies than those seen in the SWITCHMRK study populations. In the NEFA study,33 in which previous treatment was not restricted and all patients were switched to a new drug class, most patients with virological failure had received suboptimum regimens in the past, resembling the SWITCHMRK findings. More recently, highly treatment-experienced patients suppressed on combination regimens containing twice-daily injections of enfuvirtide were randomly assigned to remain on enfuvirtide or to switch to raltegravir while continuing their background regimen.34-35 In an open-label study, raltegravir was non-inferior to enfuvirtide as measured by the cumulative proportion of patients with confirmed vRNA concentration 400 copies per mL or more at 24 weeks.36
Limitations of our studies include the non-uniformity of the nucleoside and nucleotide drug regimens and the failure to account upfront for past virological failures. Although our exploratory analyses provide a plausible explanation for the SWITCHMRK findings, these inferences will need to be confirmed in prospective studies.
The SWITCHMRK studies showed that switching from lopinavir-ritonavir to raltegravir was associated with greater reductions in serum lipid concentrations than was continuation of lopinavir-ritonavir in HIV-infected patients with stable viral suppression on lopinavir-ritonavir-based combination therapy. In the heterogeneous population enrolled in the studies, a higher rate of HIV suppression was achieved in patients who continued lopinavir-ritonavir than in patients who switched to raltegravir, especially in patients who had virological failure before entry. Virological rebound in patients assigned to raltegravir was usually associated with the development of mutations associated with raltegravir resistance. By contrast, patients without previous virological failure had similar viral suppression rates in both treatment groups. Because participants were not stratified before randomisation by the subcategories used in the post-hoc analyses, imbalances in potentially important covariates might confound interpretation of these data. In practice, clinicians need to gather all available background information, including past resistance tests and treatment outcomes, when contemplating the potential risks and benefits of modifying a suppressive antiretroviral regimen. Efficacy, tolerability, and safety data from rigorous clinical trials that can be applied contextually to individual patients will help to inform these difficult decisions.
Results
Figure 1 shows the trial profiles for the SWITCHMRK 1 and 2 studies. 702 patients received at least one dose of study drug and were included in the efficacy and safety analyses (raltegravir, n=350; lopinavir-ritonavir, n=352), of whom 249 (35%) were non-white and 152 (22%) were women. Table 1 shows baseline characteristics of patients. In the combined analysis of the two studies, patients in both treatment groups had received a median of five antiretroviral agents (IQR 4-7) before study entry over a median duration of 3·4 years (2·0-7·3) in the raltegravir group and 4·1 years (2·1-7·4) in the lopinavir-ritonavir group. 112 (32%) patients in the raltegravir group and 123 (35%) patients in the lopinavir-ritonavir group had virological failure on a previous antiretroviral regimen. 38 (5%) patients (raltegravir, n=17; lopinavir-ritonavir, n=21) had vRNA concentration 50 copies per mL or more at baseline (median 100·5 copies per mL [IQR 66·0-193·0]) despite screening for concentrations less than 50 copies per mL before randomisation. 35 (10%) patients assigned to raltegravir and 23 (7%) assigned to lopinavir-ritonavir discontinued the study (figure 1). The median time in the study ranged from 37 weeks to 41 weeks across treatment groups.
In both studies at week 12, raltegravir regimens were associated with significantly greater percentage reductions in baseline fasting total cholesterol, non-HDL cholesterol, and triglycerides than were lopinavir-ritonavir regimens (p<0·0001 for each pairwise comparison; figure 2). In the combined analysis, the changes in lipid concentrations for the raltegravir group compared with the lopinavir-ritonavir group were -12·6% versus 1·0% for total cholesterol, -15·0% versus 2·6% for non-HDL cholesterol, and -42·2% versus 6·2% for triglycerides. Changes in LDL cholesterol (1·1% vs 1·2%) and HDL cholesterol (-0·7% vs -1·0%) were similar in the raltegravir and lopinavir-ritonavir groups.
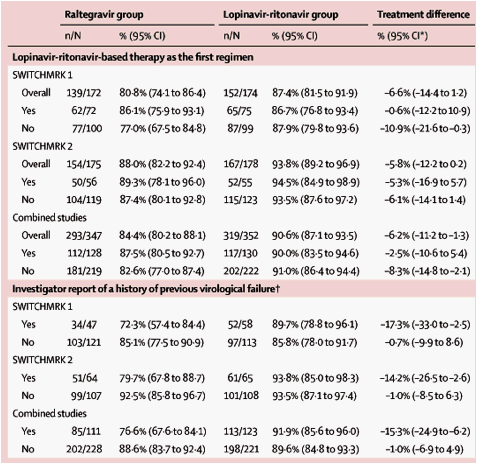
In an analysis that judged all treated patients who did not complete the study as failures, viral suppression to less than 50 vRNA copies per mL at week 24 was achieved by 139 (80·8%) patients in the raltegravir group compared with 152 (87·4%) in the lopinavir-ritonavir group in SWITCHMRK 1, and by 154 (88·0%) patients in the raltegravir group compared with 167 (93·8%) in the lopinavir-ritonavir group in SWITCHMRK 2 (figure 3). The treatment differences were -6·6% (95% CI -14·4 to 1·2) for SWITCHMRK 1 and -5·8% (-12·2 to 0·2) for SWITCHMRK 2 (table 2). In the combined analysis, viral suppression to less than 50 copies per mL was achieved by 293 (84·4%) patients in the raltegravir group compared with 319 (90·6%) patients in the lopinavir-ritonavir group at week 24, (difference -6·2%, -11·2 to -1·3). Similar results were obtained in sensitivity analyses with an observed-failure approach to missing data (data not shown).
The analysis of time to confirmed virological failure included all available data up to the date when the last patient completed the visit at week 24. Time to confirmed virological failure was similar for the raltegravir and lopinavir-ritonavir groups in SWITCHMRK 1 (log-rank test, p=0·5023), whereas it was shorter for patients in the raltegravir group than for those in the lopinavir-ritonavir group in SWITCHMRK 2 (log-rank test, p=0·0136; figure 3). Of 38 patients with baseline vRNA concentration more than 50 copies per mL, seven (41%) of 17 in the raltegravir group and ten (48%) of 21 in the lopinavir-ritonavir group had a vRNA concentration more than 50 copies per mL at week 24. Mean changes in CD4-cell count from baseline to week 24 were small, ranging from 5 cells per µL to 17 cells per µL, and did not differ between treatment groups.
Table 2 compares virological results at week 24; results are shown in patients for whom the lopinavir-ritonavir-based regimen at study entry was their first antiretroviral regimen versus patients who had received previous antiretroviral treatment, and in patients with a history of virological failure on a previous antiretroviral regimen versus those without such a history. In the subgroup of patients from both studies for whom the lopinavir-ritonavir-based regimen at study entry was their first antiretroviral regimen, the between-treatment group difference was -2·5% (95% CI -10·6 to 5·4) at week 24 (regarding all patients who did not complete the study as failures); in the subgroup of patients without history of virological failure, the between-treatment group difference was -1·0% (-6·9 to 4·9) at week 24. Similar results were seen in the sensitivity analyses with an observed-failure approach to missing data (data not shown). The differences in response rates between treatment groups in the prespecified subgroup analyses by age, ethnic origin, sex, region, hepatitis B and C status, and duration of lopinavir-ritonavir use before study entry were generally similar to the overall results (data not shown).
Clinical and laboratory adverse events occurred at similar frequencies in both treatment groups (table 3). There were no serious drug-related adverse events or deaths. In the combined analysis, the only drug-related clinical adverse event of moderate to severe intensity reported in 1% or more of either treatment group was diarrhoea, which occurred in ten patients in the lopinavir-ritonavir group (3%) and no patients in the raltegravir group. Six patients in the raltegravir group discontinued treatment early because of adverse events (hypersensitivity, n=1; diarrhoea, n=1; acute stress disorder, n=1; adverse drug reaction, n=1; raised serum concentration of alanine aminotransferase, n=2). In the lopinavir-ritonavir group, four patients discontinued treatment because of adverse events (vomiting, n=1; upper abdominal pain with diarrhoea, n=1; pulmonary tuberculosis, n=1; diarrhoea associated with an increased serum concentration of creatinine, n=1). Grade 3 or 4 laboratory abnormalities were infrequent and generally balanced between groups (table 4).
49 patients met the protocol definition of confirmed virological failure. Of 32 patients assigned to raltegravir with confirmed virological failure, 27 (84%) reported that their lopinavir-ritonavir regimen at study entry was not their first antiretroviral regimen. 18 (67%) of these patients had a history of virological failure on previous regimens. Of 17 patients assigned to lopinavir-ritonavir with confirmed virological failure, eight (47%) reported that their lopinavir-ritonavir regimen at study entry was not their first antiretroviral regimen and four (50%) of these patients reported a history of virological failure on previous regimens. Genotypic resistance testing was done on 14 of 16 patients with confirmed virological failure and vRNA concentration more than 400 copies per mL (table 5). Of the 11 assessable patients who rebounded on raltegravir-based therapy, virus with mutations known to confer raltegravir resistance was found in eight patients; in five of these eight patients, resistance mutations were also found in the reverse transcriptase gene. One additional patient had a mixture of viruses showing T97T/A, which might represent a polymorphism in the integrase gene; T97A confers a low-level decrease in susceptibility to raltegravir.
Methods
Study design
The SWITCHMRK 1 and 2 studies (MK-0518 protocols 032 and 033, respectively) were identically designed, double-blind, randomised, active-controlled clinical trials. Patients were enrolled from 81 centres in five continents between June 11, 2007, and May 15, 2008. The protocols were approved by the institutional review boards or ethics review committees at each study site. All participants provided written informed consent.
Patients with HIV infection who were aged 18 years or older were eligible for the studies if they had documented plasma viral RNA (vRNA) concentration lower than 50 copies per mL by PCR or lower than 75 copies per mL by branched DNA assay for at least 3 months while on a lopinavir-ritonavir-based regimen. The current regimen had to include at least two nucleoside or nucleotide reverse transcriptase inhibitors and no other protease inhibitors. The lopinavir-ritonavir-based regimen at study entry was not required to be the patient's first-ever antiretroviral therapy. Patients who had virological failure on previous regimens but then achieved viral suppression on a lopinavir-ritonavir-based regimen for at least 3 months were eligible. Exclusion criteria were pregnancy, breastfeeding, treatment with lipid-lowering agents during the preceding 12 weeks, or a history of diabetes mellitus, coronary artery disease, acute or decompensated chronic hepatitis, renal insufficiency requiring dialysis, or any medical disorder likely to interfere with the execution or interpretation of the study. Patients with stable chronic hepatitis were eligible if their serum aminotransferase concentrations were less than or equal to five times the upper limit of the normal range.
Randomisation and masking
Once patients had satisfied all eligibility requirements and were stratified by duration of lopinavir-ritonavir use before study entry (≤1 year vs >1 year), study site personnel accessed a central interactive voice response system (IVRS) and allocated patients according to a computer-generated randomised allocation schedule in a 1:1 ratio to receive raltegravir or lopinavir-ritonavir, each in combination with the other antiretroviral agents in their baseline regimen. Drugs were packaged, assigned unique kit numbers, and aligned with a treatment group in the IVRS database. The IVRS selected a kit number associated with the patient's treatment assignment from the inventory at the study site. Treatment allocation was concealed from investigators, study site personnel, patients, monitors, and central laboratory personnel by use of blinded access codes. Masking was maintained through use of placebo tablets with identical appearance. In a double-dummy design, participants received a 400 mg tablet of raltegravir (Isentress; Merck, Whitehouse Station, NJ, USA) or identical placebo and two lopinavir-ritonavir 200 mg/50 mg tablets (Kaletra; Abbott, Chicago, IL, USA) or identical placebos twice daily approximately 12 h apart without regard to food intake. Other antiretroviral drugs were continued without change unless modifications were needed because of toxic effects.
Procedures
vRNA concentrations were measured at a central laboratory by the Ultrasensitive Amplicor HIV-1 Monitor assay version 1.5 (Roche Diagnostics; Branchburg, NJ, USA) with a low quantification limit of 50 vRNA copies per mL. Confirmed virological failure was operationally defined as a vRNA concentration 50 copies per mL or more on two consecutive measurements at least 1 week apart. Raltegravir resistance was investigated by integrase genotyping of virus from patients who had virological failure with vRNA concentration more than 400 copies per mL.19 Other resistance testing was done on volumes of the samples from the same timepoint by the PhenoSense GT assay (Monogram Biosciences; San Francisco, CA, USA).
Primary endpoints were the mean percentage change in lipid concentrations from baseline to week 12, the proportion of patients with vRNA concentration less than 50 copies per mL at week 24, and the frequency of adverse events up to 24 weeks.
Statistical analysis
Unless otherwise noted, all analyses were specified by protocol and done with SAS software version 9.1/8.2. All randomised patients treated with at least one dose of study drug were included in the efficacy and safety analyses. Analyses of changes in lipid concentrations from baseline to week 12, and efficacy and safety analyses at week 24 were prespecified per protocol for each individual study for hypothesis testing. Analyses of the combined studies were done to provide more precise estimates of treatment effects; no hypothesis testing was specified on the composite data set in the data analysis plan.
The percentage changes from baseline in fasting LDL cholesterol, HDL cholesterol, non-HDL cholesterol, and total cholesterol at week 12 were analysed with ANCOVA models with terms for baseline lipid concentration, duration of lopinavir-ritonavir-based regimen (≤1 year vs >1 year), and treatment assignment. The percentage change from baseline in fasting triglycerides was analysed with a non-parametric ANCOVA model with terms for baseline concentration, duration of lopinavir-ritonavir-based regimen, and treatment assignment. With 170 patients in each treatment group, the individual studies had more than 99% power to detect a between-treatment difference of 11%, 53%, and 13% in the mean percentage change from baseline in total cholesterol, triglycerides, and non-HDL cholesterol, respectively, and 71% power to detect a 4% treatment difference in the mean percentage change from baseline in LDL cholesterol.
In each study, raltegravir would be judged non-inferior to lopinavir-ritonavir if the lower bound of the two-sided 95% CI for the proportion of patients with vRNA concentration less than 50 copies per mL at week 24 in the raltegravir group minus the response rate in the lopinavir-ritonavir group was higher than -12%. 95% CIs for the treatment difference were calculated by the method of Miettinen and Nurminen.21 On the assumption of a true response rate of 87·5% at week 24 for both the raltegravir and lopinavir-ritonavir groups with 170 patients in each group, the individual studies would have 90% power to show non-inferiority.
To calculate virological response rates, the primary approach for handling missing data was to regard all treated patients who did not complete the study as failures. In this analysis, missing vRNA measurements were imputed as failures, irrespective of the reason for absence, unless the values immediately before and after the missing value were both less than 50 copies per mL, in which case the absent value was recorded as missing. Sensitivity analyses with an observed-failure approach to missing data were also specified, in which no imputation was made for missing values unless the data were missing as a result of discontinuation because of lack of efficacy or the last value at discontinuation was a failure. The time to confirmed virological failure was defined as the time from randomisation until the first of two consecutive vRNA concentrations of 50 copies per mL or more measured at least 1 week apart. Kaplan-Meier estimates of time to virological failure were calculated by treatment group. Changes from baseline in CD4-cell counts were summarised over the course of the study by use of the data-as-observed method with no imputation for missing values.
Subgroup efficacy analyses were prespecified for age, ethnic origin, sex, region, hepatitis B and C status, and duration of lopinavir-ritonavir use before study entry (≤1 year vs >1 year). After reviewing the results from the protocol-specified hypotheses, we undertook exploratory analyses based upon retrospective collection of supplementary data from the two studies to examine virological response rates in patients for whom the lopinavir-ritonavir-based regimen was their first versus later antiretroviral regimen and in patients with a history of virological failure on a previous antiretroviral regimen versus those without.
Adverse events occurring at any time during the study or within 14 days after discontinuation were included in the safety analysis. Investigators assessed whether each adverse event was related to any drug in the study regimen. Investigators graded clinical adverse events as mild, moderate, or severe. Severity of laboratory abnormalities was graded according to the 1992 Division of AIDS (DAIDS) toxicity guidelines for adults.22 Adverse event terms were adopted from the Medical Dictionary for Regulatory Activities (MedDRA version 11.0).23 Frequencies of adverse events were not adjusted for duration of follow-up. The proportions of patients with clinical and laboratory adverse events were compared between treatment groups for the following categories: (1) at least one adverse event; (2) drug-related adverse events; (3) serious adverse events; (4) serious drug-related adverse events; and (5) discontinuation of study treatment because of an adverse event. For adverse events occurring in 20% of patients, each study had 80% power to declare with 95% confidence that the true difference between treatment groups was 12% or lower. If a particular adverse event was not recorded in 170 patients, it could be concluded with 95% confidence that its true incidence was 2% or lower.
A scientific advisory committee periodically reviewed blinded safety and efficacy results from each study. After examining data from the planned efficacy analyses at week 24, a recommendation to stop the studies was made because of lower than expected efficacy in patients who switched to a raltegravir-based regimen compared with patients who remained on a lopinavir-ritonavir-based regimen. These trials are registered with ClinicalTrials.gov, numbers NCT00443703 and NCT00443729.
Role of the funding source
The study was designed, managed, and analysed by the sponsor in conjunction with external investigators. Employees of the sponsor were involved in the writing of the report. The report underwent formal review by the sponsor. Authors had access to all study data upon request. The sponsor committed to publishing these data at the inception of the trial. The authors made the final decisions about when and where to publish the data.
|
|
| |
| |
|
|
|