| |
Early Immune Senescence in HIV Disease
|
| |
| |
Seema Desai1 and Alan Landay1
Department of Immunology/Microbiology, Rush University Medical Center, 1735 W. Harrison Street, Room 660 Cohn, Chicago, IL 60612, USA
"Activation and inflammation due to persistent infection such as HIV also provide a milieu for accelerated replicative senescence of T cells that progressively accumulate during the normal course of aging.....aging in the post-HAART era with suppressed viral replication is an outcome of activation and inflammation, and is most accurately defined as "inflamm-aging"....Whether HIV alone drives immunosenescence or if there are alternative pathways that contribute to early aging in HIV-infected individuals also remains to be examined.....more than 99% of HIV-1 particles detected in the circulation are not productively infectious virions. These noninfectious particles contribute to HIV-induced immunopathogenesis, as they activate the innate and adaptive immune system to release mediators of inflammation that are known to be associated with age-associated co-morbidities. The proof of this concept comes from data from the Strategies for Management of Antiretroviral Therapy (SMART) study, which shows elevated levels of tumor necrosis factor-α (TNF-α), interleukin-1ß (IL-1ß), and IL-6 to be associated with non-AIDS-defining co-morbidities in HAART-suppressed patients. The persistence of HIV virions, infectious or noninfectious, in the circulation results in the constant stimulation of the immune system and likely drives early senescence in HIV infection.....alterations in immune homeostatic mechanisms may lead to progressive loss of the naïve and memory T-cell pool, resulting in an imbalance in T-cell phenotypes. Altered T-cell homeostasis impairs regulatory cell function. HIV may deplete regulatory CD4+ T cells, which are normally responsible for suppressing T-cell activation and limiting the amount of inflammatory damage to tissues. Excessive production and/or accumulation of proinflammatory mediators such as TNF-α, IL-1ß, and IL-6 in HIV infection and in the elderly suggests that immune activation coupled with lack of anti-inflammatory responses likely results in accelerated aging in HIV disease....direct activation of innate immune cells by HIV and by disruption of the gastrointestinal barrier due to HIV-mediated depletion of Th-17 cells, leading to microbial translocation, could be contributing factors to activation and inflammation in the accelerated aging process."
Current HIV/AIDS Reports
Published online: 2 February 2010
Abstract
Non-AIDS-defining co-morbidities that occur despite viral suppression and immune reconstitution using antiretroviral therapy depict early aging process in HIV-infected individuals. During aging, a reduction in T-cell renewal, together with a progressive enrichment of terminally differentiated T cells, translates into a general decline of the immune system, gradually leading to immunosenescence. Inflammation is a hallmark of age-associated comorbidities, and immune activation is a hallmark of HIV disease. Constant stimulation of the immune system by HIV or due to co-infections activates the innate and adaptive immune system, resulting in release of mediators of inflammation. Immune activation coupled with lack of anti-inflammatory responses likely results in accelerated aging in HIV disease. Dysfunctional thymic output, along with HIV-mediated disruption of the gastrointestinal barrier leading to microbial translocation, contributes to the circulating antigenic load driving early senescence in HIV disease.
Introduction
Now that we are more than 25 years into the HIV epidemic, there are many more HIV-infected individuals of older age due to improvements in antiretroviral therapy. From 2000 to 2004, the Centers for Disease Control reported that the proportion of AIDS patients who are ≥50 years of age rose from 19% to 27% and that the number of adults ≥50 years of age living with HIV infection and/or AIDS more than doubled. Importantly, for that surveillance period, persons 40 to 49 years of age had the highest prevalence of HIV/AIDS and the steepest rise in prevalence. The number of older people with HIV/AIDS is expected to increase even further during the next decade. It is projected that by 2015, more than half of all HIV-infected individuals in the United States will be over the age of 50 years [1]. Adults with HIV on prolonged treatment with highly active antiretroviral therapy (HAART) frequently experience long-term side effects of disease and treatment that mimic natural aging processes (Fig. 1).
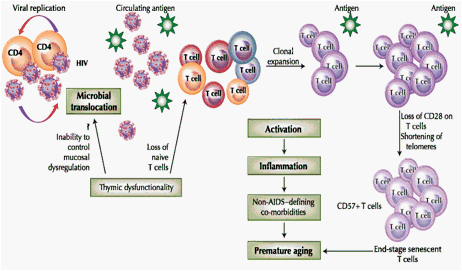
Fig. 1 Accelerated aging model in HIV infection. Viral replication results in release of virions (infectious and noninfectious HIV) into circulation. Residual ongoing replication continues to activate immune cells despite highly active antiretroviral therapy. Microbial translocation adds to the antigenic burden. Loss of thymic function alters T-cell homeostasis. Immune activation due to circulating antigen is the central event in the senescent pathway. Activated cells undergo clonal expansion in response to the persistent antigen, resulting in differentiation and accumulation of nonfunctional end stage senescent cells. Activated cells release inflammatory mediators, causing optimal and suboptimal inflammation associated with non-AIDS-defining co-morbidities and premature aging
Increasing evidence suggests that HIV-1-infected individuals experience similar immunologic changes as uninfected elderly persons. An increasing number of investigators have reported osteoporosis [2], atherosclerosis [3], and neurocognitive decline [4] in HIV-1-infected patients, and HIV-1 disease progression is also associated with onset of frailty usually related to old age [5]. Thus, physiologic alterations and co-morbidities suggest that advanced aging occurs in HIV disease. Several immunologic alterations that characterize HIV-1-infected individuals are remarkably similar to those associated with age in the HIV-1-uninfected elderly. During aging, a reduction in T-cell renewal, together with a progressive enrichment of terminally differentiated T cells with shortened telomeres, occurs. It is thought that these changes are a consequence of immune activation and inflammation, which translates into a general decline of the immune system, gradually leading to immunosenescence (aging of the immune system) [6]. This article examines accelerated aging in HIV disease as an activation-induced inflammatory condition that is a consequence of optimal or suboptimal inflammation and activation due to antigen- (infectious or noninfectious HIV) driven injury that occurs over the lifetime, rather than just a complex group of diseases or morbidities associated with age or HIV infection. The use of an integrated strategy to control activation and inflammation rather than treat individual diseases is likely the modality to control advanced aging in the HIV-infected individual.
Immune activation is a hallmark of chronic HIV infection. Immune activation occurs despite effective HIV control with HAART and is a critical factor contributing to HIV pathogenesis [7]. Activation and inflammation due to persistent infection such as HIV also provide a milieu for accelerated replicative senescence of T cells that progressively accumulate during the normal course of aging [8]. HIV infects CD4 T cells, the profound depletion of which results in immunodeficiency and terminal AIDS. Immune activation is postulated to be the leading cause associated with non-AIDS-defining co-morbidities [9]. Whether these non-AIDS-defining co-morbidities would occur despite the control of ongoing HIV replication with antiretroviral therapy or whether they are an outcome of an aging immune system is currently under investigation. Whether HIV alone drives immunosenescence or if there are alternative pathways that contribute to early aging in HIV-infected individuals also remains to be examined. First and foremost, more than 99% of HIV-1 particles detected in the circulation are not productively infectious virions [10]. These noninfectious particles contribute to HIV-induced immunopathogenesis, as they activate the innate [11] and adaptive [12] immune system to release mediators of inflammation that are known to be associated with age-associated co-morbidities. The proof of this concept comes from data from the Strategies for Management of Antiretroviral Therapy (SMART) study, which shows elevated levels of tumor necrosis factor-α (TNF-α), interleukin-1ß (IL-1ß), and IL-6 to be associated with non-AIDS-defining co-morbidities in HAART-suppressed patients [13]. The persistence of HIV virions, infectious or noninfectious, in the circulation results in the constant stimulation of the immune system and likely drives early senescence in HIV infection.
Secondly, alterations in immune homeostatic mechanisms may lead to progressive loss of the naïve and memory T-cell pool, resulting in an imbalance in T-cell phenotypes. Altered T-cell homeostasis impairs regulatory cell function. HIV may deplete regulatory CD4+ T cells, which are normally responsible for suppressing T-cell activation and limiting the amount of inflammatory damage to tissues [14]. Excessive production and/or accumulation of proinflammatory mediators such as TNF-α, IL-1ß, and IL-6 in HIV infection and in the elderly suggests that immune activation coupled with lack of anti-inflammatory responses likely results in accelerated aging in HIV disease. Finally, direct activation of innate immune cells by HIV and by disruption of the gastrointestinal barrier due to HIV-mediated depletion of Th-17 cells, leading to microbial translocation, could be contributing factors to activation and inflammation in the accelerated aging process.
Cause and Consequence of Early Senescence in HIV Disease
Persistent Activation and Inflammation Accelerates Aging in HIV Disease
Among untreated patients with HIV infection, higher percentages of CD8+ T cells expressing the activation marker CD38 predicts rapid clinical progression to AIDS and death more strongly than CD4+ T-cell counts and plasma HIV RNA levels [15]. Activation is more pronounced in the CD8+ T-cell subset compared with the CD4+ T-cell subset, which is known to be susceptible to phenotypic changes in aging elderly as well [16]. Accumulation of CD8+ T cells in HIV-infected and aging individuals suggests that activation of these cells may contribute inflammatory mediators. Cells of the innate system (eg, monocytes, dendritic, and natural killer [NK] cells) are also activated by infectious and noninfectious HIV particles and secrete inflammatory cytokines such as TNF-α, IL-1ß, and IL-6 that cause tissue damage in a manner similar to aging processes [8]. Immune activation and inflammation are associated with chronic HIV infection and are also likely to be the cause of systemic aging of physiologic functions [8]. A recent study suggests that an increase in peripheral proinflammatory Th-17 cells and a decrease in anti-inflammatory regulatory T cells was associated with acute coronary disease and arthrosclerosis [17]. A recent report in murine model indicates a 20-fold increase in IL-17 and a threefold increase in IL-6 production in splenic CD4+ T cells from 22- to 24-month-old C57BL/6 and CBA mice compared with mice 6 to 10 weeks of age [18]. Prior studies in atherosclerosis and Alzheimer's disease reported a relationship between cytokine polymorphisms and longevity, suggesting that those individuals who were genetically predisposed to produce low levels of inflammatory cytokines (IL-6 and TNF-α) or high levels of anti-inflammatory cytokines (IL-10) may have an increased capacity to reach the extreme limit of human life-span [19]. These studies provide insights into inflammatory mechanisms that result in accelerated aging in HIV disease. Excessive production of proinflammatory mediators such as TNF-α, IL-1ß, and IL-6 with lack of anti-inflammatory response likely results in accelerated aging in HIV disease. Thus, aging in the post-HAART era with suppressed viral replication is an outcome of activation and inflammation, and is most accurately defined as "inflamm-aging" [20].
Altered T-Cell Homeostasis as a Consequence of Early Immunosenescence
Failure of adaptive immunity with age is a major cause of morbidity and mortality in the elderly. Thymic production of new T cells dwindles with age and does not meet replenishment demands during adulthood. The capacity of the thymus to produce new cells is also significantly reduced in aging individuals. Several factors may account for this decline of thymic output, such as the direct infection of the thymic stroma and thymocytes by viruses (cytomegalovirus being the most common age-associated virus infection), and the atrophy of thymus may be related to thymosuppressive effects of proinflammatory cytokines (such as IL-6) [21]. After age 50 years, virtually the entire T-cell supply is generated from existing naïve and memory T cells. An insufficient homeostatic mechanism may lead to progressive loss of the naïve and memory T-cell pool. HIV alters the fragile homeostatic control of T-cell subsets. Continuous stimulation of the immune system in HIV disease with loss of CD4+ T cells by activation-induced cell death coupled with poor T-cell restoration [22] due to lower thymic function results in an imbalance in T-cell phenotypes and is similar to that observed in the elderly [23]. As a consequence, the naïve T-cell pool cannot be replenished efficiently, and is therefore unable to replace old senescent CD8+ T-cell clones and depleted CD4+ T cells in HIV-infected individuals, leading to impaired T-cell homeostasis.
Levels of cellular activation are a major driving factor of proliferation and T-cell differentiation, resulting in the generation of antigen-experienced cells that eventually lose expression of CD28 and increase the expression of CD57, which is a key predictor of immune incompetence in the elderly and HIV-infected individuals. These subpopulations tend to lose the capacity to produce IL-2 and demonstrate a decline in proliferative capacity that is associated with a shortening of telomere lengths, so that highly differentiated cells (CD28-/CD57+) have been considered T cells that are approaching end stage senescence. CD28 is a co-stimulatory molecule on T cells, and upon binding to its ligand CD80 on antigen-presenting cells, transduces downstream survival and proliferation signals, including induction of IL-2 and its receptor, telomerase activation, stabilization of several cytokine mRNAs, as well as providing essential survival signals for T cells [24]. The loss of CD28 compromises both B- and T-cell responses. Loss of CD28 on CD4+ T cells alters its capacity to help B-cell proliferation and antibody production, thus affecting vaccine responses in elderly. Accumulation of memory T cells and clonal expansion results in restricted T-cell diversity [25]. Thus, loss of CD28 on T cells is a key predictor of immune aging. A recent study reported that the CD57 expression on HIV-1-specific CD8+ T cells defines the proliferative defects after antigenic stimulation in vitro better than the lack of CD28 expression. An elevated proportion of CD8+ T cells expressing CD57 has been observed in both aging [26] and HIV-1 infection [27].
HIV-1 infection induces premature aging of naive CD4+ T cells and memory CD4+ and CD8+ T cells. A recent study reported that premature aging of T cells was associated with faster HIV-1 disease progression [28]. In HIV-1-infected individuals, the majority of CD8+ T cells were CD28-, as compared with less than 10% within the CD4+ T-cell subset, similar to phenotypic changes in aging [28]. The differences in susceptibility of both T-cell subsets to aging can be attributed to differences in kinetics of loss of CD28 upon repeated antigenic stimulation or their differences in life-span [16, 29]. Therefore, despite a significant number of CD8+ T cells observed during HIV-1 infection, the CD8+ T-cell compartment is mostly comprised of functionally defective CD28- T cells. The mechanism for the accelerated loss of CD28 expression during HIV-1 infection remains to be elucidated. Their accumulation during HIV-1 infection may be a combined result of accelerated antigen-driven differentiation and prolonged survival due to the apoptosis resistance property ascribed to this CD8+ T-cell subset [25]. Some researchers report that the accumulation of CD28-CD8+ T cells is associated with a decline of CD4+ T cell counts [30] or AIDS development [31], whereas others have suggested that CD28 expression on CD4+ T cells but not CD8+ T cells is independently associated with disease progression [32].
Our own results [33] suggest that HAART-treated patients with good immune reconstitution and suppressed viral load below detectable levels had a phenotype similar to older HIV-negative individuals. T cells had higher immune activation and senescence levels and reduced naïve and central memory subsets as compared to younger HIV-negative individuals. We compared HIV-infected HAART-suppressed (<80 RNA copies/mL, median CD4 counts of 724 cells/mm3) patients with older HIV-negative individuals (median age, 88 years) and HIV-negative young controls (median age, 27 years). We found significantly higher frequency of senescent CD8+ T cells (CD57+CD28-) in HIV-infected and older HIV-negative individuals as compared to HIV-negative young controls. The degree of CD8+ T cell activation (HLADR+CD38+) was significantly higher in HIV-infected patients as compared to HIV-negative older and younger controls. Naïve CD4+ and CD8+ T cells (CD45RA+CCR7+) were significantly reduced in HIV- infected patients and older HIV-negative individuals as compared to HIV-negative young controls, with proportional increase in terminally differentiated effector T cells (CD45RA+CCR7-). CD4+ and CD8+ central memory T cells (CD45RA-CCR7+) were significantly reduced in HIV- infected individuals as compared to older HIV-negative individuals. Thus, altered T-cell homeostasis, immune activation, and senescence are hallmarks of early aging in HIV infection, and as our results suggest, HIV-infected individuals (median age, 56 years) with good immune reconstitution and viral suppression had immune changes comparable with older (median age, 88 years) HIV-uninfected individuals.
Microbial Translocation: A Cause or Consequence of Premature Aging in HIV Disease
Is senescence antigen driven or does it have a parallel detrimental pathway due to secondary effects of immune dysregulation? Chronic immune activation likely accelerates senescence that is driven by HIV itself or by co-infections (CMV, hepatitis B virus, hepatitis C virus, etc). How the dysfunctional thymus contributes to chronic immune activation that accelerates immunosenescence and whether microbial translocation is the cause or consequence of early senescence are a few of the questions that are addressed here. Recent studies in mice have shown that ablation of the thymus and loss of naïve T cells results in immune activation of CD4+ T cells, which correlated with higher microbial load in the periphery, indicating that continued replenishment with cells from the thymus seems to be required to maintain efficient gut mucosal defenses [34].
Using a model of chemical thymectomy to study peripheral T-cell homeostasis by inducible Rag ablation, Bourgeois et al. [34] reported a rapid decay of naïve T cells and decay of maintenance of memory and regulatory T cells. The decay of naïve CD4 T cells was more pronounced than CD8 T cells. They attribute this decay of naïve CD4+ T cells as compared with naïve CD8+ T cells to changes in the lymphopenic environment, resulting in higher microbial load and activation and not to intrinsic survival differences of the two T-cell subsets. The authors convincingly show the involvement of gut-derived antigens in the activation of naïve CD4 T cells by demonstrating higher concentrations of lipopolysaccharide binding protein in lymphopenic Rag-deficient mice as compared to wild-type mice. Their results clearly indicate that CD4 T cells react to a changed environment, whereas naïve CD8 T cells do not seem to perceive any environmental changes and are not activated. This highlights the fact that thymic export in some way minimizes and corrects adverse effects of peripheral activation of naïve CD4 T cells by environmental/gut-derived stimuli. Activated CD4 T cells do not control the antigenic load, suggesting that new supply of naïve CD4 T cells from the thymus can guarantee effective control of gut-derived antigens. These findings suggest that in lymphopenic conditions, such as aging, and in chronic viral infections, such as HIV, that survival of a naïve CD4 T-cell repertoire may be severely curtailed because of chronic activation and also suggest that thymic output seems to be required to maintain efficient gut mucosal defense.
The effects of aging on gut-associated lymphoid tissue (GALT) are poorly understood, but clinical evidence of increased infections due to organisms in which gastrointestinal immunity plays a vital role (eg, Clostridium difficile colitis [35]) suggests that there are likely to be age-related changes in GALT as well. HIV infection is associated with CD4+ T-cell depletion, which predominantly occurs in the GALT. Although the mechanism of CD4+ T-cell depletion is partially due to direct effects of viral replication, it is known that CD4+ T-cell depletion also occurs via activation-induced cell death. Preferentially lost in the gut are Th-17 cells that constitute host defense mechanisms against bacterial infections. Massive depletion of Th-17 cell in the gut results in loss of barrier integrity, causing gut leakiness and translocation of microbes and microbial products into circulation as observed by elevated bacterial lipopolysaccharide [36]. In HIV infection, microbial translocation is known to be associated with chronic immune activation [37]. From the murine study by Bourgeois et al. [34], it appears that augmenting thymic output is a major consideration to curtail aberrant activation caused by HIV infection or HIV-induced microbial translocation. Thus, microbial translocation is an outcome of HIV pathology, and an aging immune system fails to control gut-derived antigens due to dysfunctional thymic output, suggesting that HIV infection is the cause and premature aging the consequence of microbial translocation.
Aging Innate Immune Response in HIV
Similar to the decline in adaptive immune function, the function of NK cells, monocytes, macrophages, dendritic cells, and neutrophils are crucial cellular components of innate immunity that are functionally impaired with aging [38]. Impairment in sensing machinery in innate cells occurs with aging. Murine studies suggest that macrophages from aged mice respond poorly to ligands for TLR1-5 and 9 and secrete significantly lower levels of IL-6 and TNF-α compared with young mice. These results support the concept that increased susceptibility to infections and poor adaptive immune responses in aging may be due to the decline in Toll-like receptor (TLR) expression and function [39]. On the contrary, in other studies tissue samples from senescent animals demonstrate elevated levels of activated nuclear factor-κB [18, 40]. Nuclear factor-κB is a major regulator of innate immunity, and its activation via TLR stimulation results in the expression of genes encoding acute phase proteins and in the production and secretion of proinflammatory cytokines. Thus, age-associated inflammation and TLR dysfunction appear to be paradoxical. Some explanation for this paradox could be that age-associated inflammation occurs in uninfected aged animals, which is a very different physiologic condition than during infection. A recent murine study examined whether aging and chronic inflammation and TLR dysfunction were associated with increased susceptibility to infection with Streptococcus pneumoniae [41]. This study reported that aged mice and young mice infused with TNF-α were more susceptible to infection with Streptococcus pneumoniae and that aged mice had lower TLR dysfunction with proinflammatory cytokine production. Thus, preexisting inflammation could prime susceptibility to infection, and age-dependent TLR dysfunction could result in a delayed and/or muted immune response. In humans too, TLR dysfunction can predispose aging individuals to infections [42]. These findings can be extrapolated to viral infections. Aging impairs both TLR7 and TLR9 responses in plasmacytoid dendritic cells (pDC), the innate sensors that mediate interferon-α (IFN-α) response to viral infections. A murine study conducted to elucidate whether aging impairs activation of pDC though TLR9 reported impaired viral clearance due to decreased IFN-α production [43]. Aging leads to increased oxidative stress in pDC and decreased upregulation of interferon regulatory factor-7 (IRF-7), an important molecule in IFN-α signaling pathway. Reducing oxidative stress in aged pDCs partly recovers the age-induced IFN-α defect during TLR9 activation [43].
Paradoxically, IFN-α in HIV infection is known to contribute to immune activation of CD8 T cells [44]. Age-associated defects in TLR recognition and in HIV infection could compromise host defenses to viral clearance, augmenting the persistence of antigen in circulation and driving senescence. Thus, increased susceptibility or persistence of infection is primed by the very mediators of inflammation and activation that are required for their elimination/clearance, contributing to age-associated morbidities.
Controlling Early Senescence in HIV Infection
HAART is the best immune deactivator and enables the reduction of naïve T-cell consumption and helps to restore their numbers, although restoration is known to be suboptimal. Given the fact that low level of viral replication occurs below the detectable threshold in HAART-treated individuals, adding another drug to current regimens (such as the integrase inhibitor) to improve outcomes is currently under investigation [45]. Other strategies may be developed to block or minimize immune activation and inflammation. These could include the use of immunosuppressive drugs (eg, cyclosporine A), inhibitors of bacterial product-mediated effects (eg, antagonists of TLR-4), the receptor for lipopolysaccharide [46], or inhibitors of proinflammatory cytokines (eg, anti-IL-1ß, anti-IL-6, or anti-TNF-α) [47]. Adjuvant therapies such as r-hIL-7 that stimulate recent thymic emigrant have not only shown impressive, rapid increases in CD4+ T cell count, but also improves the number of CD4+ central memory cells and naïve T cells, deficits of which are associated with immune dysfunction in many patients treated long term as well as being associated with accelerated senescence [48]. Based on the central role of telomere shortening in the replicative senescence, several telomerase-based approaches are currently being tested as potential immunoenhancing treatments for aging and HIV disease. CD8+ T cells are unable to upregulate telomerase upon repeated antigenic stimulation. It has been shown in vitro that gene therapy of HIV-specific CD8+ T cells with the telomerase catalytic component (hTERT) results in enhanced proliferative capacity, increased antiviral functions, and a delay in the loss of CD28 expression [49]. Most intriguing are results with a small molecule telomerase activator (TAT2) that modestly retards telomere shortening, increases proliferative potential, and enhances cytokine/chemokine production and antiviral activity in TAT-2 exposed CD8 T+ lymphocytes from HIV-infected human donors [50].
Conclusions
Activation and inflammation occur in both HIV infection and aging without HIV infection, and both conditions share a common detrimental pathway that leads to early immune senescence. This includes direct activation of adaptive and innate immune cells by infectious and noninfectious HIV antigens and indirect induction of activation and inflammation by HIV disruption of the gastrointestinal barrier and subsequent microbial translocation. Thus, accelerated aging with HIV is indeed the "plague" of this post-HAART decade, and much of this early senescence is related to inflammation and chronic immune activation. Where and how to target activation-induced inflammation should be the focus (now that there exists enough evidence that this phenomenon occurs) for better quality of life for those afflicted by this malady.
Acknowledgement
Jules Levin, Executive Director and Founder, NATAP for driving our attention to important work done and to be done on aging and HIV research.
Disclosure No potential conflicts of interest relevant to this article were reported.
References
1. Effros RB, Fletcher CV, Gebo K, et al.: Aging and infectious diseases: workshop on HIV infection and aging: what is known and future research directions. Clin Infect Dis 2008, 47:542-553.
2. Teichmann J, Stephan E, Discher T, et al.: Changes in calciotropic hormones and biochemical markers of bone metabolism in patients with human immunodeficiency virus infection. Metabolism 2000, 49:1134-1139.
3. Kaplan RC, Kingsley LA, Sharrett AR, et al.: Ten-year predicted coronary heart disease risk in HIV-infected men and women. Clin Infect Dis 2007, 45:1074-1081
4. Ikezu T: The aging of human-immunodeficiency-virus-associated neurocognitive disorders. J Neuroimmune Pharmacol 2009, 4:161-162.
5. Desquilbet L, Margolick JB, Fried LP, et al.: Relationship between a frailty-related phenotype and progressive deterioration of the immune system in HIV-infected men. J Acquir Immune Defic Syndr 2009, 50:299-306.
6. Pawelec G, Effros RB, Caruso C, et al.: T cells and aging (update february 1999). Front Biosci 1999, 4:D216-269.
7. Hunt PW, Brenchley J, Sinclair E, et al.: Relationship between T cell activation and CD4+ T cell count in HIV-seropositive individuals with undetectable plasma HIV RNA levels in the absence of therapy. J Infect Dis 2008, 197:126-133.
8. Appay V, Sauce D: Immune activation and inflammation in HIV-1 infection: causes and consequences. J Pathol 2008, 214:231-241.
9. Deeks SG, Phillips AN: HIV infection, antiretroviral treatment, ageing, and non-AIDS related morbidity. BMJ 2009, 338:a3172.
10. Piatak M Jr, Saag MS, Yang LC, et al.: High levels of HIV-1 in plasma during all stages of infection determined by competitive PCR. Science 1993, 259:1749-1754.
11. Hardy AW, Graham DR, Shearer GM, Herbeuval JP: HIV turns plasmacytoid dendritic cells (pDC) into TRAIL-expressing killer pDC and down-regulates HIV coreceptors by Toll-like receptor 7-induced IFN-alpha. Proc Natl Acad Sci U S A 2007, 104:17453-17458.
12. Esser MT, Bess JW, Jr., Suryanarayana K, et al.: Partial activation and induction of apoptosis in CD4(+) and CD8(+) T lymphocytes by conformationally authentic noninfectious human immunodeficiency virus type 1. J Virol 2001;75:1152-1164.
13. Kuller LH, Tracy R, Belloso W, et al.: Inflammatory and coagulation biomarkers and mortality in patients with HIV infection. PLoS Med 2008, 5:e203.
14. Eggena MP, Barugahare B, Jones N, et al.: Depletion of regulatory T cells in HIV infection is associated with immune activation. J Immunol 2005, 174:4407-4414.
15. Giorgi JV, Lyles RH, Matud JL, et al.: Predictive value of immunologic and virologic markers after long or short duration of HIV-1 infection. J Acquir Immune Defic Syndr 2002, 29:346-355.
16. Czesnikiewicz-Guzik M, Lee WW, Cui D, et al.: T cell subset-specific susceptibility to aging. Clin Immunol 2008, 127:107-118.
17. Cheng X, Yu X, Ding YJ, et al.: The Th17/Treg imbalance in patients with acute coronary syndrome. Clin Immunol 2008, 127:89-97.
18. Huang MC, Liao JJ, Bonasera S, et al.: Nuclear factor-kappaB-dependent reversal of aging-induced alterations in T cell cytokines. Faseb J 2008, 22:2142-2150.
19. Caruso C, Candore G, Colonna-Romano G, et al.: Inflammation and life-span. Science 2005, 307:208-209; author reply 208-209.
20. Ginaldi L, De Martinis M, Monti D, Franceschi C: Chronic antigenic load and apoptosis in immunosenescence. Trends Immunol 2005, 26:79-84.
21. Linton PJ, Dorshkind K: Age-related changes in lymphocyte development and function. Nat Immunol 2004, 5:133-139.
22. Kelley CF, Kitchen CM, Hunt PW, et al.: Incomplete peripheral CD4+ cell count restoration in HIV-infected patients receiving long-term antiretroviral treatment. Clin Infect Dis 2009, 48:787-794.
23. Douek DC, McFarland RD, Keiser PH, et al.: Changes in thymic function with age and during the treatment of HIV infection. Nature 1998, 396:690-695.
24. Lenschow DJ, Walunas TL, Bluestone JA: CD28/B7 system of T cell costimulation. Annu Rev Immunol 1996, 14:233-258.
25. Spaulding C, Guo W, Effros RB: Resistance to apoptosis in human CD8+ T cells that reach replicative senescence after multiple rounds of antigen-specific proliferation. Exp Gerontol 1999, 34:633-644.
26. Merino J, Martinez-Gonzalez MA, Rubio M, et al.: Progressive decrease of CD8high+ CD28+ CD57- cells with ageing. Clin Exp Immunol 1998, 112:48-51.
27. Brenchley JM, Karandikar NJ, Betts MR, et al.: Expression of CD57 defines replicative senescence and antigen-induced apoptotic death of CD8+ T cells. Blood 2003, 101:2711-2720.
28. Cao W, Jamieson BD, Hultin LE, et al.: Premature aging of T cells is associated with faster HIV-1 disease progression. J Acquir Immune Defic Syndr 2009, 50:137-147.
29. Valenzuela HF, Effros RB: Divergent telomerase and CD28 expression patterns in human CD4 and CD8 T cells following repeated encounters with the same antigenic stimulus. Clin Immunol 2002, 105:117-125.
30. Gamberg J, Pardoe I, Bowmer MI, et al.: Lack of CD28 expression on HIV-specific cytotoxic T lymphocytes is associated with disease progression. Immunol Cell Biol 2004, 82:38-46.
31. Choremi-Papadopoulou H, Panagiotou N, Samouilidou E, et al.: CD28 costimulation and CD28 expression in T lymphocyte subsets in HIV-1 infection with and without progression to AIDS. Clin Exp Immunol 2000, 119:499-506.
32. Choi BS, Park YK and Lee JS. The CD28/HLA-DR expressions on CD4+T but not CD8+T cells are significant predictors for progression to AIDS. Clin Exp Immunol 2002, 127:137-144.
33. Desai SR, Usuga X, Martinson J, et al.: Immune senescence, activation and abnormal T cell homeostasis despite effective HAART, a hallmark of early aging in HIV. Presented at 16th Conference on Retroviruses and Opportunistic Infections. Montreal; February 8-11, 2009.
34. Bourgeois C, Hao Z, Rajewsky K, et al.: Ablation of thymic export causes accelerated decay of naïve CD4 T cells in the periphery because of activation by environmental antigen. Proc Natl Acad Sci U S A 2008, 105:8691-8696.
35. Redelings MD, Sorvillo F, Mascola L: Increase in Clostridium difficile-related mortality rates, United States, 1999-2004. Emerg Infect Dis 2007, 13:1417-1419.
36. Cohen J: Retrovirus meeting. Back-to-basics push as HIV prevention struggles. Science 2008, 319:888.
37. Brenchley JM, Price DA, Schacker TW, et al.: Microbial translocation is a cause of systemic immune activation in chronic HIV infection. Nat Med 2006, 12:1365-1371.
38. Panda A, Arjona A, Sapey E, et al.: Human innate immunosenescence: causes and consequences for immunity in old age. Trends Immunol 2009, 30:325-333.
39. Renshaw M, Rockwell J, Engleman C, et al.: Cutting edge: impaired Toll-like receptor expression and function in aging. J Immunol 2002, 169:4697-4701.
40. Helenius M, Hanninen M, Lehtinen SK, Salminen A: Changes associated with aging and replicative senescence in the regulation of transcription factor nuclear factor-kappa B. Biochem J 1996, 318:603-608.
41. Hinojosa E, Boyd AR, Orihuela CJ: Age-associated inflammation and toll-like receptor dysfunction prime the lungs for pneumococcal pneumonia. J Infect Dis 2009, 200:546-554.
42. van Duin D, Mohanty S, Thomas V, et al.: Age-associated defect in human TLR-1/2 function. J Immunol 2007, 178:970-975.
43. Stout-Delgado HW, Yang X, Walker WE, et al.: Aging impairs IFN regulatory factor 7 up-regulation in plasmacytoid dendritic cells during TLR9 activation. J Immunol 2008, 181:6747-6756.
44. Martinson J, Montoya CJ, Usuga X, et al.: Chloroquine modulates HIV-1 induced plasmacytoid dendritic cell IFNα: implication for T cell activation. Antimicrob Agent Chemother 2010, 54(2):871-881.
45. Hatano H, Delwart EL, Norris PJ, et al.: Evidence for persistent low-level viremia in individuals who control human immunodeficiency virus in the absence of antiretroviral therapy. J Virol 2009, 83:329-335.
46. Czeslick E, Struppert A, Simm A, Sablotzki A: E5564 (Eritoran) inhibits lipopolysaccharide-induced cytokine production in human blood monocytes. Inflamm Res 2006, 55:511-515.
47. Connolly NC, Riddler SA, Rinaldo CR: Proinflammatory cytokines in HIV disease-a review and rationale for new therapeutic approaches. AIDS Rev 2005, 7:168-180.
48. Levy Y, Lacabaratz C, Weiss L, et al.: Enhanced T cell recovery in HIV-1-infected adults through IL-7 treatment. J Clin Invest 2009, 119:997-1007.
49. Dagarag M, Evazyan T, Rao N, Effros RB: Genetic manipulation of telomerase in HIV-specific CD8+ T cells: enhanced antiviral functions accompany the increased proliferative potential and telomere length stabilization. J Immunol 2004, 173:6303-6311.
50. Fauce SR, Jamieson BD, Chin AC, et al.: Telomerase-based pharmacologic enhancement of antiviral function of human CD8+ T lymphocytes. J Immunol 2008, 181:7400-7406.
| |
| |
| |
|
|
|