| |
ANA-598 HCV Non-Nuke Phase 2b Study Preliminary Data Results
Background to Phase IIb Study
|
| |
| |
Anadys laid the foundation for the current Phase IIb clinical trial with prior clinical and preclinical work. In a Phase IIa combination trial in HCV patients, we reported data that showed that setrobuvir added to pegylated interferon and ribavirin accelerated the rate of viral clearance, with comparable response at setrobuvir doses of 200 mg bid and 400 mg bid. A single patient out of more than 60 exhibited viral breakthrough while receiving setrobuvir plus standard of care, corresponding to a low breakthrough rate of < 2%. Setrobuvir also showed an excellent safety profile in the study through the 12 weeks of dosing, with reported adverse events being typical for patients treated with interferon and ribavirin alone, although conclusions regarding safety cannot be made until results in more patients over longer duration are known.
In a Phase I trial in HCV patients, setrobuvir demonstrated potent antiviral activity when dosed as monotherapy over three days. No patient at any dose level in the monotherapy study showed evidence of viral breakthrough while on setrobuvir, and there were no serious adverse events reported. Anadys has presented in vitro data supporting the use of setrobuvir in combination with interferon-alpha as well as several DAAs currently in development that act through diverse mechanisms. Data has shown that setrobuvir is synergistic in vitro with interferon-alpha as well as with representative HCV protease and polymerase inhibitors. In vitro combination treatment at clinically relevant concentrations of interferon-alpha and setrobuvir results in clearance of HCV RNA from cells rather than selection of resistant isolates. Furthermore, setrobuvir retains full activity in vitro against mutations conferring resistance to protease inhibitors, nucleoside polymerase inhibitors and non-nucleoside polymerase inhibitors that act at binding sites distinct from that of setrobuvir, while protease and nucleoside polymerase inhibitors retain full activity in vitro against mutations conferring resistance to setrobuvir.
In 2009 Anadys completed long-term, chronic toxicology studies of setrobuvir, of 26 weeks duration in rats and 39 weeks duration in monkeys, and has reported favorable results that supported dosing for as long as a year in clinical studies.
Preclinical evaluation of setrobuvir was completed in the first quarter of 2008, leading to submission of an Investigational New Drug Application (IND) to the FDA, subsequent allowance of the IND by the FDA and initiation of clinical investigation in the second quarter of 2008.
ANADYS ANNOUNCES POSITIVE 12-WEEK DATA
FOR SETROBUVIR IN PHASE 2B HEPATITIS C STUDY
· Strong Antiviral Response in Prior Partial Responders and Relapsers
· Favorable Safety Data with AE Profile Comparable to Control Group
· High Barrier to Resistance Confirmed
Conference Call at 8:00 AM EDT Today
SAN DIEGO, October 13, 2011 -- Anadys Pharmaceuticals, Inc. (Nasdaq: ANDS) today released interim antiviral response and safety data from an ongoing Phase IIb study of setrobuvir in combination with pegylated interferon and ribavirin (P/R) in genotype 1 hepatitis C patients. Setrobuvir is the Company's direct-acting antiviral being developed for the treatment of chronic hepatitis C, or HCV.
"We are pleased with today's data, which we believe demonstrate a compelling profile for setrobuvir in significantly more patients," said Steve Worland, Ph.D., President and CEO of Anadys. "The antiviral response in patients who had failed prior treatment is a particularly encouraging benchmark of setrobuvir's potency and high barrier to resistance. Coupled with a favorable safety profile to date, we believe today's data position setrobuvir as a very attractive agent to be included in future DAA combination regimens."
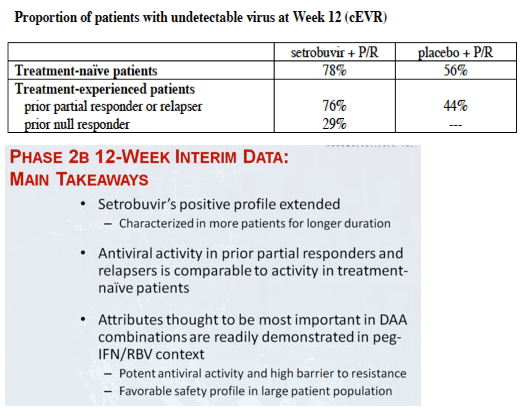
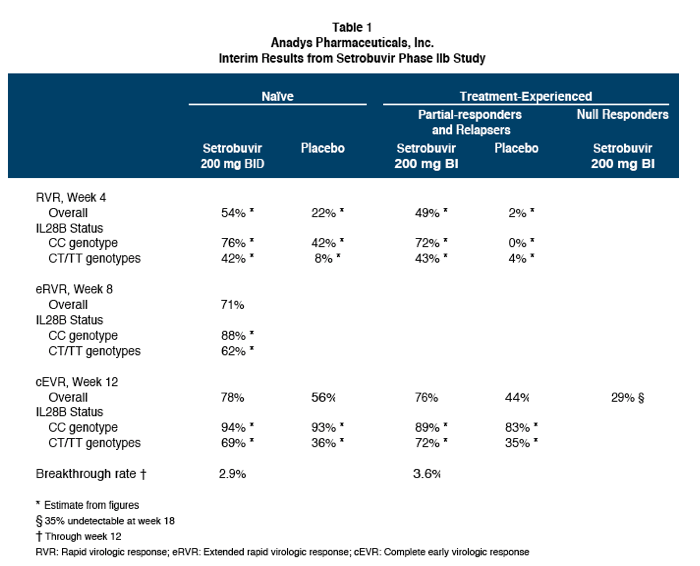
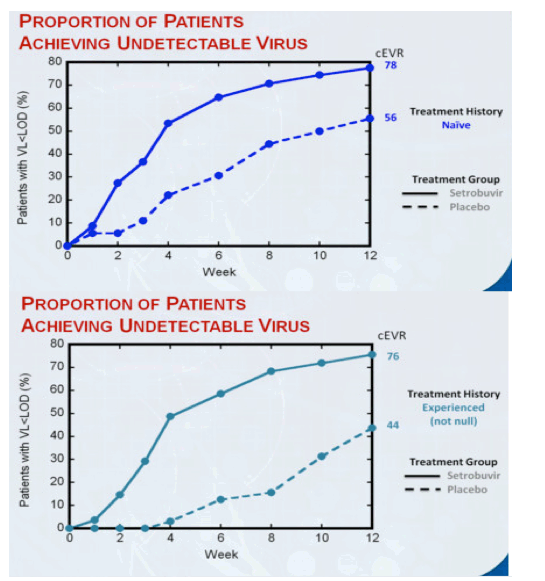
78% of treatment-naïve patients and 76% of patients who had responded inadequately to, or relapsed after, prior treatment with P/R had undetectable virus at week 12 (cEVR) while receiving setrobuvir plus P/R, compared to 56% and 44%, respectively, for patients who received placebo plus P/R. 71% of treatment-naïve patients who received setrobuvir plus P/R had undetectable virus at week 8 (from Jules: why week 8 instead of week 4 like telaprevir? slow response to 598) and met the initial response-guided criteria for shortening treatment in this study to 28 weeks from the traditional 48 weeks for treatment with P/R alone. 29% of patients who had no appreciable response to prior treatment with P/R (null responders) achieved cEVR with setrobuvir plus P/R, and the percentage of patients with undetectable virus continued to climb in this hard-to-treat population to 36% at week 18. No prior null responders received placebo plus P/R in this trial.
"In experienced patients: response rates in partial responders and relapsers were comparable to those seen in naïve patients; cEVR rate is 29% in null responders. In prior partial-responders and relapsers, setrobuvir+P/R demonstrated cEVR and RVR rates of 76% and approximately 49%, respectively, versus 44% and 2% for placebo+P/R...... Low breakthrough continues to make setrobuvir a very special non-nuke......." 71% naïve patients met the early stop criteria at week 8
The viral breakthrough rate through 12 weeks on setrobuvir plus P/R was low in both treatmentnaïve patients (2.9%) and patients who had responded inadequately to, or relapsed after, prior treatment with P/R (3.6%). The Company believes this low incidence of viral breakthrough exhibited to date in a larger patient population further characterizes setrobuvir's high barrier to resistance.
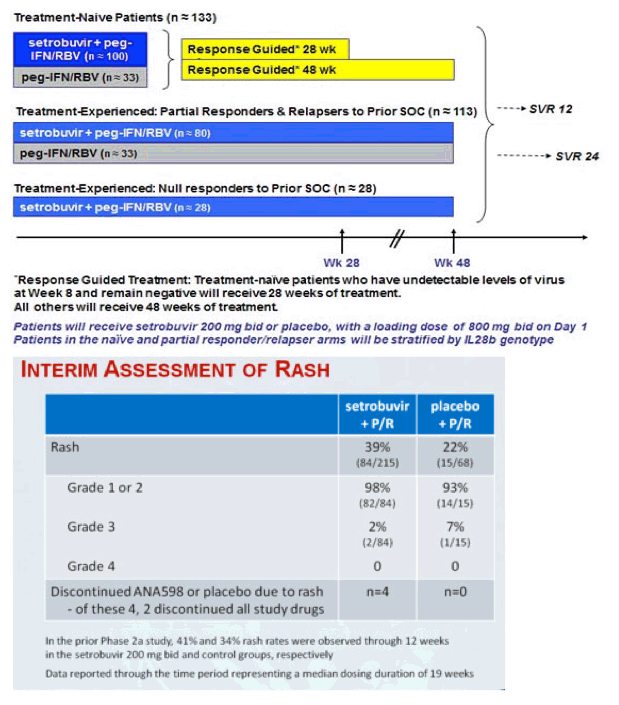
Setrobuvir has been generally well-tolerated in the study. Safety data for patients receiving setrobuvir plus P/R, and comparison to the control group that received placebo plus P/R, are being reported through a time period that reflects a median dosing duration of 19 weeks. The rate of discontinuing treatment in the study due to adverse events has been similar in patients receiving setrobuvir plus P/R (5.6%) or P/R alone (5.9%). The profile of adverse events has been similar between the setrobuvir and control groups, with reported adverse events being typical for patients treated with interferon and ribavirin. In the setrobuvir group, 39% of patients (84/215) developed a rash while 22% (15/68) of patients in the control group developed a rash. 98% of the rashes in the setrobuvir group were mild or moderate (grade 1 or grade 2), compared to 93% in the control group. There were no Grade 4 rashes in either group. The incidence of rash in the setrobuvir group is consistent with prior reports of rash due to interferon and ribavirin through 19 weeks of treatment.
Phase 2b Protocol Design
283 patients were dosed in this study. Patients received either setrobuvir 200 mg twice a day (bid) in combination with Pegasys® (peginterferon alfa-2a) and Copegus® (ribavirin, USP) (P/R) with a loading dose of setrobuvir 800 mg bid on day 1, or placebo plus P/R. Patients who had a prior null response to P/R, defined as less than a 1 log10 decline in viral load at week 4 or less than a 2 log10 decline at week 12 during prior treatment, were not randomized to receive placebo.
All other patients were stratified by IL28B genotype (CC/non-CC) between setrobuvir and placebo groups. The interim antiviral response data is being reported as the proportion of patients with undetectable virus (< 15 IU/mL) using the Roche COBAS HCV TaqMan assay. The primary endpoint of the study is Sustained Virological Response 24 weeks after patients conclude all treatment, known as SVR24. In addition to the interim data released today, data through 24 weeks of dosing are expected around year-end. The study is being conducted at sites in the United States, Canada, Australia and New Zealand.
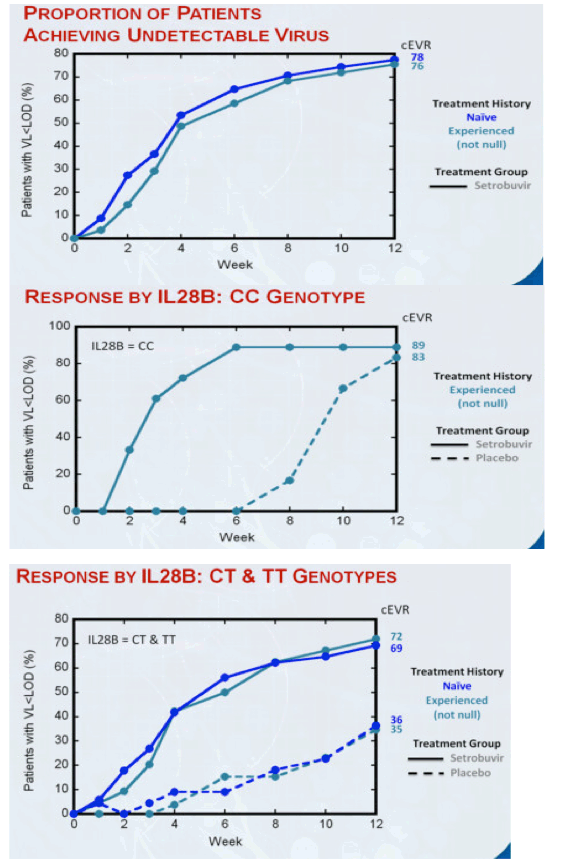
Treatment-Naïve Group
· 102 treatment-naïve patients received setrobuvir plus P/R
· 36 treatment-naïve patients received placebo plus P/R
· Treatment-naïve patients who achieved undetectable levels of virus by Week 8 and maintain undetectable levels of virus are scheduled to conclude all treatment at Week 28
· For treatment-naïve patients with detectable virus at Week 8, treatment with setrobuvir or placebo and P/R is scheduled to continue through Week 48
Treatment-Experienced Group (including prior null responders)
· 82 patients who were partial responders during, or relapsers after, a prior course of therapy with P/R alone received setrobuvir plus P/R
· 32 corresponding patients received placebo plus P/R
· 31 prior null responder patients received setrobuvir plus P/R
· All treatment-experienced patients are scheduled to be treated for 48 weeks
About Setrobuvir
Setrobuvir is an HCV RNA polymerase inhibitor that belongs to a chemical class referred to as non-nucleosides. Setrobuvir has a well-characterized safety database in which more than 350 subjects have received the agent to date. Setrobuvir has received Fast Track Status from the FDA for the treatment of chronic hepatitis C. Earlier this year, Anadys announced a cross-company clinical trial agreement with a large, commercial-stage biopharmaceutical company to study setrobuvir in combination with another DAA in healthy volunteers.
|
|
| |
| |
|
|
|