| |
Boceprevir Case Study
|
| |
| |
Download the PDF here
Hepatology december 2011
Haripriya Maddur and Paul Y. Kwo
From the Gastroenterology/Hepatology Division, Indiana University School of
Medicine, Indianapolis, IN
Received October 24, 2011; accepted October 25, 2011.
Address reprint requests to: Paul Y. Kwo, M.D.,
Gastroenterology/Hepatology
Division, Indiana University School of Medicine, 975 West Walnut, IB 327,
Indianapolis, IN 46202-5121. E-mail: pkwo@iupui.edu; fax: 317-274-3106
Boc, boceprevir; HCV, hepatitis C virus; IL-28, interleukin-28; PCR, polymerase chain reaction; PR, pegylated interferon and ribavirin; SPRINT-2, Serine Protease Inhibitor Therapy 2; SVR, sustained virological response.
Case Scenario
A 52-year-old male executive who is asymptomatic is evaluated for abnormal liver biochemical tests. The aspartate aminotransferase level is 138 U/L, and the alanine aminotransferase level is 164 U/L; the bilirubin, alkaline phosphatase, and albumin levels and the complete blood counts are normal. The international normalized ratio is 1.1, and the serum creatinine level is 0.9 mg/dL. The hepatitis C virus (HCV) RNA level is 1,600,000 IU/mL, and the genotype is 1B. The patient has read about boceprevir and wants to know whether he is a candidate for treatment with this drug. He also wants to know whether he really requires liver biopsy before the initiation of treatment.
Will you use boceprevir in this patient? How will you determine whether he is responding to the drug, how long will you give him the medication, and how will you monitor him for side effects? How will you determine that treatment-related anemia is related to boceprevir and is not related to ribavirin? Which side effects of boceprevir will warrant the discontinuation of treatment? Will your approach vary with the genotype for the interleukin-28 (IL-28) polymorphism?
The Problem
Chronic HCV affects approximately 170 million people worldwide.1 HCV, the most common blood-borne infection in the United States, is a major cause of chronic liver disease, which can lead to death from liver failure or hepatocellular carcinoma.2-4 For the past decade, therapy for HCV infection has entailed the use of pegylated interferon and ribavirin (PR). Although the sustained virological response (SVR) rates with this treatment regimen have been as high as 80% for genotypes 2 and 3, the rates for genotype 1 have been less favorable (approximately 40%-50%).4-6 In May 2011, the Food and Drug Administration approved two direct-acting antiviral agents, telaprevir and boceprevir, for the treatment of HCV genotype 1 in both previously untreated patients and patients who failed to achieve SVR with PR.7 When they are added to the standard of care (PR), SVR rates for genotype 1 infections are markedly improved in patients who have not been treated; SVR rates of 63% to 75% have recently been reported.8, 9 Boceprevir is not currently recommended for HCV genotype 2 or 3 infections.
Boceprevir
The current treatment regimens with direct-acting antiviral agents incorporate nonstructural protein 3 protease inhibitors in conjunction with PR. Boceprevir is a linear peptidomimetic keto amide serine protease inhibitor that binds reversibly to the HCV nonstructural protein 3 active site.10 In the recently reported Serine Protease Inhibitor Therapy 2 (SPRINT-2) study,10 the addition of boceprevir to pegylated interferon alfa-2b and ribavirin significantly improved SVR rates in nonblack patients from 38% for patients with just PR to 67% for patients with 24 weeks of boceprevir in response-guided treatment arms (28 or 48 weeks of therapy according to viral clearance). Patients who received 48 weeks of treatment (i.e., 44 weeks of boceprevir with PR after a 4-week PR lead-in period) achieved an SVR rate of 68% (Supporting Fig. 1).
The side effects associated with the addition of boceprevir to PR include increased rates of dysgeusia, neutropenia, and anemia. Dysgeusia that is observed when boceprevir is added to the standard of care is usually mild and rarely, if ever, requires the discontinuation of therapy. Although neutropenia may lead to infections in those receiving PR, severe infections are infrequent, and treatment cessation is rarely warranted. Anemia associated with triple therapy (PR and boceprevir) is primarily driven by ribavirin-related hemolytic anemia, which begins during the 4-week PR lead-in period and is responsible for the majority of the hemoglobin decline.11 Anemia associated with boceprevir typically contributes an additional decline of 1 g/dL to the decline associated with ribavirin therapy. Anemia associated with boceprevir is thought to be due to the bone marrow-suppressive effect of the drug, whereas anemia associated with ribavirin is attributed to hemolysis. Similar to the development of anemia with PR therapy, the development of anemia with boceprevir-based treatment is associated with higher SVR rates.12 In the SPRINT-2 trial, dose modifications due to anemia were required almost twice as often for patients on boceprevir regimens versus the PR control groups (21% versus 13%). However, the rates of discontinuation due to adverse events were not significantly different for the patients on boceprevir-containing regimens (13%) and the PR controls (12%), and discontinuation due to anemia was rare as well (2% for the patients on boceprevir-containing regimens and 1% for the PR controls). It should be emphasized that erythropoietin supplementation was used in the trial.
Drug interactions are significant with boceprevir and are discussed in the next section.
Monitoring for Drug-Related Side Effects
Boceprevir is primarily metabolized by two pathways: the aldo-keto reductase pathway and the cytochrome P450 3A4 pathway. Importantly, it is a reversible inhibitor of cytochrome P450 3A4. All individuals who are candidates for boceprevir therapy require an assessment of drug-drug interactions (Supporting Table 1).
Before therapy is started, thyroid-stimulating hormone levels must be determined, and pregnancy testing is required for women of child-bearing potential. Additionally, complete blood count monitoring should be performed before treatment initiation, at weeks 2, 4, 6, 8, and 12, and monthly thereafter.
The management of anemia involves ribavirin dose reductions (200 mg/day) when hemoglobin levels decline to <10 g/dL in patients without underlying cardiovascular disease or when there is a >2 g/dL drop in hemoglobin levels over any 4-week interval during the treatment course (including the 4-week lead-in period). If the hemoglobin concentration declines to <8.5 g/dL, ribavirin should be discontinued. The boceprevir dose should never be reduced during ribavirin or interferon dose modifications. Additionally, if the discontinuation of either interferon or ribavirin is required, all three treatments should be discontinued to prevent potential boceprevir resistance.
Areas of Uncertainty
Need for Liver Biopsy Before Treatment.
A liver biopsy sample provides important information about the prognosis and the urgency of treatment and excludes other forms of liver disease.13 The degree of fibrosis has also been shown to be an independent predictor of the response to therapy. In patients with HCV genotype 1 infections, the need for liver biopsy is less compelling because of the higher SVR rates observed with the addition of boceprevir to the standard of care. However, information about advanced fibrosis from a pretreatment liver biopsy sample may be used to predict the response to therapy, even with the advent of newer direct-acting antiviral agents. Indeed, in the SPRINT-2 study, the SVR rates of patients with F3/F4 fibrosis in the boceprevir arms were only 41% to 52%. If a patient's liver biopsy sample reveals mild fibrosis (F0-F2), there is a higher chance of SVR (67% in the SPRINT-2 study) with boceprevir-based treatment. A finding of minimal fibrosis may reduce the urgency of therapy, and the patient could await possible newer therapies. On the other hand, if the liver biopsy sample demonstrates cirrhosis, 48 weeks of treatment is recommended.
Testing for IL-28 Polymorphisms.
The SVR rates for PR-treated HCV genotype 1 patients with the IL-28 CC genotype were more than 2-fold greater than the rates for patients with the CT or TT genotype.14-15 Data regarding the use of IL-28B with the addition of direct-acting antiviral agents to PR are emerging, and as the discovery of IL-28B occurred after the large phase 3 trials with telaprevir and boceprevir had been initiated, we will need to wait for more complete data sets in naive patients. In the SPRINT-2 trial, IL-28 data were available for 62% of the patients (653/1048). The addition of boceprevir was associated with higher SVR rates for the patients with the IL-28 CT and TT genotypes (Supporting Fig. 1).16 Those with the IL-28 CC genotype had SVR rates comparable to those of the controls, but 88% of these individuals cleared the virus by week 8 and were eligible for short-term (28-week) therapy.
Testing for IL-28 polymorphisms could be used for counseling patients. If a patient has the IL-28 CC genotype, he may require only 28 weeks of therapy instead of 48 weeks. If he has the IL-28 CT or TT genotype, the addition of boceprevir will substantially improve his chances of SVR in comparison with just PR therapy. However, we could also use a week 4 viral decline after the PR lead-in period as a marker because an HCV decline at 4 weeks appears to be a stronger predictor of SVR than the IL-28 status.
Use of Erythropoiesis-Stimulating Agents.
In the SPRINT-2 study, 43% of the patients receiving boceprevir-based therapy received erythropoiesis-stimulating agents, whereas only 24% of the PR-receiving controls did. Thromboembolic events have been associated with the use of erythropoiesis-stimulating agents in patients with peginterferon-alfa-treated HCV, and these agents are not approved for the treatment of ribavirin-related anemia. In the SPRINT-2 study, similar SVR rates were observed regardless of the anemia management strategies, which included ribavirin dose reductions, erythropoiesis-stimulating agents, both ribavirin dose reductions and erythropoiesis-stimulating agents, and no dose modifications.12 These findings call into question the precise role of erythropoiesis-stimulating agents when antiviral agents are used for the treatment of HCV. A large, prospective, randomized trial evaluating the use of an erythropoiesis-stimulating agent versus ribavirin dose reduction in patients receiving boceprevir with PR is fully enrolled (ClinicalTrials.gov identifier: NCT01023035) and should address this important question.
Recommendations
This patient is clearly a candidate for therapy with boceprevir and PR and has a high possibility of achieving SVR. Liver biopsy, although it is not required, may help with prognostication. IL-28 testing may be helpful, especially if the patient is interested in truncating therapy with no compromise in the chance of achieving SVR. The treatment will entail a 4-week lead-in period with PR alone and then the addition of boceprevir (800 mg every 7-9 hours) with a light meal or snack, and his viral load response during the treatment will determine the treatment duration. The viral load can be reduced during the lead-in period before the addition of boceprevir, and this period can be used to assess the responsiveness to interferon/ribavirin and to predict the likelihood of SVR resistance. Indeed, if the viral decline is >1 log10 at the end of week 4, this patient has a >80% chance of achieving SVR with a response-guided treatment paradigm (Supporting Table 1). However, if the viral decline during the lead is <1 log10, then he is poorly responsive to interferon and will require PR and boceprevir for 44 weeks. HCV RNA levels should be determined at weeks 4, 8, 12, and 24 of therapy and at the end of the treatment course. When HCV RNA is undetectable by polymerase chain reaction (PCR) at weeks 8 and 24 of treatment with assay with lower limit of detection (LLOD) of 10-15 IU/ml (weeks 4 and 20 of boceprevir therapy), the treatment can be completed after 28 weeks (Fig. 1). If, however, HCV RNA is detectable by PCR at week 8 but is undetectable at week 24 (with PCR test with LOD <10-15 IU/ml), treatment with boceprevir should be continued until week 36 and should be followed by PR alone until week 48. The continuation of therapy despite these parameters will lead to a marked increase in the risk of resistance-associated variants.
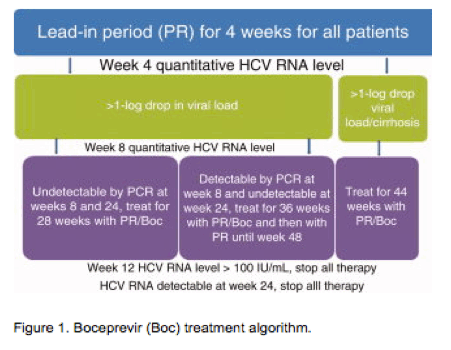
Careful management of anemia will be required, but the preliminary data suggest that the anemia management strategy will not affect SVR rates. Finally, the measurement of viral levels at weeks 4, 8, 12, and 24 and adherence to futility rules will maximize SVR rates and minimize the emergence of resistance-associated variants.
Boceprevir is marketed in the United States as Victrelis by Merck. It is supplied as oral capsules at a strength of 200 mg. The cost for 24 weeks of boceprevir is approximately $25,000, and the cost for 44 weeks of therapy is approximately $46,000. The total cost of 28 weeks of triple therapy (including boceprevir) is $55,000, and the total cost of 48 weeks of therapy is approximately $101,000.
|
|
| |
| |
|
|
|