| |
Telaprevir (Incivek brand name): Previously Treated Adults Study Results
|
| |
| |
Study C216 (REALIZE)
Study C216 was a randomized, double-blind, placebo-controlled, trial conducted in subjects who did not achieve SVR with prior treatment with Peg-IFNalfa-2a/RBV or Peg-IFN-alfa-2b/RBV. The study enrolled prior relapsers (subjects with HCV-RNA undetectable at end of treatment with a pegylated
interferon-based regimen, but HCV RNA detectable within 24 weeks of treatment follow-up) and prior non-responders (subjects who did not have
undetectable HCV-RNA levels during or at the end of a prior course of at least 12 weeks of treatment). The nonresponder population included 2 subgroups:
prior partial responders (greater than or equal to 2-log10 reduction in HCV-RNA at week 12, but not achieving HCV RNA undetectable at end of treatment
with peginterferon alfa and ribavirin) and prior null responders (less than 2-log10 reduction in HCV-RNA at week 12 of prior treatment with peginterferon
alfa and ribavirin).
Subjects were randomized in a 2:2:1 ratio to one of two INCIVEK combination treatment groups (with and without a Peg-IFN-alfa-2a/RBV lead-in) or a
control group. The T12/PR48 group received INCIVEK and Peg-IFN-alfa-2a/RBV for 12 weeks (without a lead-in), followed by placebo and Peg-IFN-alfa-
2a/RBV for 4 weeks, followed by Peg-IFN-alfa-2a/RBV for 32 weeks. The T12(DS)/PR48 group had a lead-in (delayed start of INCIVEK) with placebo and
Peg-IFN-alfa-2a/RBV for 4 weeks, followed by INCIVEK and Peg-IFN-alfa-2a/RBV for 12 weeks, followed by Peg-IFN-alfa-2a/RBV for 32 weeks. The
Pbo/PR48 group received placebo and Peg-IFN-alfa-2a/RBV for 16 weeks, followed by Peg-IFN-alfa-2a/RBV for 32 weeks.
The 662 enrolled subjects had a median age of 51 years (range: 21 to 70); 70% of the subjects were male; 26% had a body mass index greater than or equal to 30 kg/m2; 5% were Black; 11% were Hispanic or Latino; 89% had baseline HCV-RNA levels greater than 800,000 IU/mL; 22% had bridging fibrosis; 26% had cirrhosis; 54% had HCV genotype 1a, and 46% had HCV genotype 1b. Null and partial responders had higher baseline HCV-RNA levels and more advanced liver disease (cirrhosis) than relapsers; other characteristics were similar across these populations.
The lead-in and immediate start regimens produced comparable SVR and no SVR rates, so data from these two groups were pooled (Table 11).
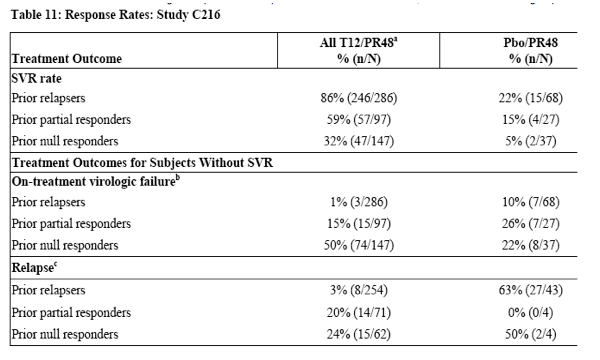
a Lead-in and immediate start T12/PR regimens pooled
b On-treatment virologic failure includes subjects who met a protocol-defined virologic stopping rule or who had detectable HCV-RNA at the time of their
last dose of INCIVEK and subjects who had viral breakthrough on peginterferon alfa/ribavirin.
C Relapse rates are calculated with a denominator of subjects with undetectable HCV-RNA at the end of treatment.
Among prior relapsers, 76% (218/286) achieved an eRVR and of those 95% (208/218) achieved an SVR. In an earlier, dose-finding clinical trial, 78%
(52/67) of prior relapsers achieved an eRVR and were treated with 24 weeks of peginterferon alfa and ribavirin (T12/PR24); of those 94% (49/52) achieved
an SVR.
For all populations in the study (prior relapsers, prior partial responders, and prior null responders), SVR rates were higher for the T12/PR group than for the
Pbo/PR48 group across subgroups by sex, age, ethnicity, body mass index, HCV genotype subtype, baseline HCV-RNA level, and extent of liver fibrosis.
Twenty-three percent of INCIVEK-treated subjects had cirrhosis at baseline. SVR rates among cirrhotic subjects who received INCIVEK combination
treatment compared to Pbo/PR48 were: 87% (48/55) compared to 13% (2/15) for prior relapsers, 34% (11/32) compared to 20% (1/5) for prior partial
responders, and 14% (7/50) compared to 10% (1/10) for prior null responders.
Four percent (19/530) of treatment experienced subjects who received INCIVEK combination treatment were Black/African Americans; the SVR rate for
these subjects was 63% (12/19) compared to 65% (328/503) for Caucasians.
|
|
| |
| |
|
|
|