| |
HIV Persistence during Therapy 5th International Workshop
(Dec 6-9 2011 St Martin, West Indies)
|
| |
| |
David Margolis MD, University of North Carolina at Chapel Hill
The Fifth International Workshop on HIV Persistence during Therapy moved back across the line to the Dutch side of the West Indies isle of St. Martin from December 6th to 9th. Hosted and organized by Alain Lafeuillade, the meeting is held in odd years, and the groundswell of energy and interest in this new area of HIV research means that a seat at this focused 250-person workshop was not casually obtained. The meeting was marked by interesting scientific reports, and diverse activity by investigative groups around the world. Again as in 2009, there was industry support for the meeting itself, and some substantial evidence of industry investment in research and development, as well as continued growth in support from the NIH and foundations such as amFAR. Clearly this area of the research is in its infancy, but there are signs of continued growth. Several of the presentations are summarized below; I believe that all abstracts will again be published in the Global Antiviral Journal (www.ihlpress.com/gaj.html).
The first morning session focused on models of HIV persistence, namely the SIV model in various non-human primates (NHP) and studies in the "humanized" mouse - a mouse model in which the immune system of a genetically immunodeficient mouse, bred to accept a human cell transplant, is reconstituted with human immune cells, and infected with HIV. Jeff Lifson (NCI-Frederick) first provided an overview of simian models of HIV persistence. He characterized an ideal NHP model was one that would support robust viremia, establish relevant reservoirs, undergo authentic pathogenesis, and respond to relevant antiretroviral therapy (ART), establishing "full" viral suppression. In the NHP field, it was not felt that fully suppressive ART, defined somewhat empirically as <50 copies of SIV RNA/ml of plasma, has yet been broadly and reproducibly established in most animals studied thus far. Dr. Lifson characterized the ongoing work in his lab and those of others as "building the boat as we sail."
Various NHPs are in use and differ in their response to SIV infection: Rhesus macaques from Indian and Chinese origin, and Pigtail macaques. Several SIV strains have been used, some with special neurotropic/neuropathogenic qualities. Chimeric viruses with an HIV RT, the RT-SHIV, can be treated with standard NNRTIs. ART poses challenges in the pharmacokinetics of drugs in NHPs, the drug delivery and cost, and toxicities that are not fully worked out. Sensitive assays of SIV RNA and DNA species by PCR in plasma, PBMC, and tissue are beginning to be worked out but are challenging. Assays of rescue of replication-competent virus have been described but are difficult. The leading studies now described suppression of viremia to 3-10 copies/ml but require the use of 5 ART drugs. Studies of ART in tissue are still needed to define if low-level viremia seen is a sign of inadequate ART or of the same persistent, low-level viremia seen in human studies. In summary, Lifson felt the field was making good advances, but was still challenged by many competing models, none of which were yet clearly superior, and the need to further optimize techniques.
Paul Luciw (UC Davis; abstract 1) reported findings of a study that attempted to deplete persistent RT-SHIV infection in macaques by the use of efavirenz, PMPA (the active form of tenofovir), and FTC in combination with intermittent administration of valproic acid and prostratin. Six weeks after infection, ART was given for 32-35 weeks, and then animals were given daily valproate and prostatin for 2 weeks, and then weekly valproate and prostratin for 6 weeks. After these cycles, ART was given for 1 week more, and then Art was stopped. There appeared to be some delay in viral rebound compared to control animals that did not receive valproate and prostatin, but this was not statistically significant. Tissue studies showed generally less SIV RNA after the receipt of valproate and prostatin, but these animals also received more weeks of ART. A follow-up study using more potent ART is planned.
Andrea Savarino (Istituto Superiore di Sanita, Rome; abstr. 2) reported a study of 18 macaques treated with multi-drug ART (tenofovir, FTC, and raltegravir, it appeared) and cycles of auranofin (reported to selectively kill memory T-cells) and the glutathione synthesis inhibitor buthionine sulfoximine (BSO), shown in tissue culture experiments to facilitate the death of HIV-infected cells. In a 2010 Retrovirology paper the group had shown suppression of viremia to < 40 copies/ml with RAL, TDF, and FTC. In 2011 in Retroviriology, the addition of auranofin to this therapy had resulted in one macaque showing control of SIV viremia after interruption of ART. Here Savarion reported that in a total of 18 macaques in different studies given different therapies, the viral setpoint after ART interruption correlated with number of ART drugs, the duration of ART, the administration of immune modulating agents such as BSO and auranofin, and the number of cycles of such agents. Later, I. Shytaj from the same group (abstr 6) reported that the addition of maraviroc to this ART regimen (Mega-ART) lowered viral load still further, and also lowered the viral rebound setpoint.
Vicente Planelles (University of Utah, abstr. 3) presented results from the system developed with Alberto Bosque in his laboratory to educate naïve T cells from the peripheral blood of healthy donors ex vivo in the presence of select cytokine cocktails and antigenic stimulation through CD3/CD28, to generate HIV-infected central memory T cells (TCM). This model system provides valuable opportunities to study how natural factors and microenvironments (such as cytokines, inflammation, stress) as well as potential anti-latency drugs affect the latent HIV reservoir. In this system they find that antigenic stimulation, as reproduced by simultaneous co-stimulation of the CD3 and CD28 receptors with binding antibodies, reactivates virtually all latently infected cells, whereas stimulation with IL-2 and IL-7 reactivates about one tenth of the latently infected population. However such cytokine stimulation induces homeostatic proliferation of TCMs. Thus proliferate of TCM in response to IL-2 and IL-7 stimulation, in the absence of viral reactivation, propagates the latent proviruses through mitosis. This may contribute to the stability and longevity of the latent reservoir.
J. Victor Garcia (UNC; abstr 4) discussed the generation of HIV Latency in BLT Humanized Mice. These mice can be fully implanted with a human immune system. BLT mice were then HIV-infected and treated with ART consisting of daily intraperitoneal injections of emtricitabine (FTC; 140-200 mg/kg), tenofovir disoproxil fumarate (TDF; 146-208 mg/kg) and raltegravir (RAL; 56-80 mg/kg). Viral load was reduced by 3.2 log to below the limit of detection in as few as eleven days. ART also dramatically reduced viral RNA levels in all the tissues examined. However, residual RNA was present in several tissues despite treatment. ART interruption led to rapid rebound of plasma viremia. It was then demonstrated that latently infected, resting CD4+ T cells are present in BLT mice and that they can be isolated with the same procedure used to obtain similar cells from human peripheral blood. The latent reservoir of HIV infected cells present in infected BLT mice on ART appears similar in frequency to that in humans 9.9 (+/- 2.4 SEM) infected resting CD4+ T cells per million human cells in the mouse. These cells are isolated primarily from tissue as well as blood. Testing of drugs to interrupt or deplete latent infection in this model is underway.
Christine Zink (abstr, 7) and Julia Russel (abstr. 5) from Johns Hopkins reported findings in an accelerated SIV CNS pigtailed macaque (Macaca nemestrina) model, treated with a four-drug antiretroviral regimen, wherein untreated animals die of NeuroAIDS within a few months. Zink said that treatment with PMPA, the integrase inhibitor L812, atazanavir, and saquinavir suppressed viremia to 3-5 copies/ml. In these animals viral envelope proteins were still found in the brain, as was HIV DNA, showing persistence. But HIV RNA in the CSF was only detected at very low levels, and it was thought that "ongoing replication" in the brain was a result of the trafficking of infected cells into the CNS. Markers of activation also persisted within the CNS, despite ART and successful suppression. Russell noted that CD68+ macrophages were found to increase over time in the SIV-infected spleen, and SIV DNA and RNA were still detected at necropsy despite ART and plasma suppression.
The next morning session focused on basic molecular mechanisms that allowed HIV persistence despite ART. Jonathan Karn (Case Western; Abstr. 8) discussed the dual roles of chromatin restriction of HIV expression and factor deficiency. During latent infections HIV proviruses are highly restricted for both transcriptional initiation and elongation. Karn explained that restrictions to HIV gene expression are first effected by modification of histones within chromatin, and then by the lack of the key positive factor pTEF-b in differentiating T cells. Overall, he suggested that some combination of approaches to remove the epigenetic histone marks that silence the provirus, restricting transcription initiation, along with signals to remove restrictions to transcriptional elongation by increasing access to P-TEFb, and/or removing restrictions enforced by NELF would likely allow the effective disruption of HIV latency.
Jose Alcami (Instituto de Salud Carlos III, Madrid; abstr. 10) discussed the cellular functions of different domains of the viral transactivator Tat. The core of Tat, amino acids 1-72, have long been known to be sufficient for viral activation. Alcami reviewed evidence that that tail domain of Tat, when present in the full-length 1-101 amino acid protein, dysregulated other cellular functions, perhaps establishing an environment more suitable to viral replication. Some of these effects included changes in cell structure, resistance to apoptosis, increased viral mRNA splicing, and activation of protein kinase C theta.
Finally, Jerry Zack (UCLA, abstr, 11) suggested approaches to clear reservoirs of persistently infected cells. He discussed the delivery to cells of the protein kinase C agonist, bryostatin, shown to induce expression of latent provirus while also downregulating receptors needed for new rounds of infection. He proposed targeting these liposomes to CD4 cells with CD4 antibodies encased in the liposomes, and in newer, unpublished work using the cellular VAULT protein to deliver bryostatin using a protein engineering approach.
As the afternoon sessions of Day 1 began, Sarah Palmer (Karolinska; abstr 12) reported single-genome and single-proviral sequencing techniques, used to obtain 20-50 single viral genomes from pretherapy plasma samples prior to the initiation of suppressive therapy from two groups of patients: 5 patients who initiated therapy during acute infection when the virus typically is homogeneous; and 3 patients who initiated therapy during chronic infection when the virus is more genetically diverse. The genetic variation and average pair-wise difference of pretherapy sequences were compared to single proviral HIV-1 genomes derived from a broad spectrum of HIV-infected cells (including naive and memory CD4+ T-cells, myeloid cells, and hematopoietic progenitor cells (HPCs)) from peripheral blood, GALT, and BMT samples of the 8 patients collected after 4-10 years of suppressive therapy. The researchers were following up on the recent claim by the Collins lab that human bone marrow progenitor cells were latently infected with HIV.
In contrast, Palmer found that Lin-/ CD34+ HPCs sorted from the bone marrow of the eight patients did not contain any HIV-1 DNA, and no HIV could be recovered in outgrowth assays from these cells. This equated with up to less than one HIV-infected cell in 800,000. In addition, no HIV-1 DNA was found in monocytes sorted from peripheral blood. Both naïve and total memory CD4+ T-cells contained HIV DNA after long-term therapy. A few myeloid cells had HIV DNA, but all of these appeared to have rearranged T cell receptors, and so were likely T cells that had undergone downregulation of receptors.
The infection frequencies of central and effector memory CD4+ T-cells from the peripheral blood and GALT were similar for both patient groups. In all 8 patients, regardless whether therapy was initiated during acute or chronic infection, phylogenetic analyses and measurements of intra-patient diversity revealed no change in viral diversity or population structure between pretherapy plasma-derived RNA sequences and intracellular DNA sequences from T cells located in the peripheral blood and GALT despite 3-12 years of suppressive therapy. Numerous intracellular HIV sequences identified after long-term therapy contained replication-incompetent virus. One patient, who initiated therapy during chronic infection, had a predominant intracellular clone in both memory and effector memory T cells containing a 380 bp deletion after > 9 years of therapy.
The absence of infected HPCs provides strong evidence that the HIV-1 infection frequency of Lin-/CD34+ HPCs from bone marrow, if it occurred, was <0.003% (highest upper 95% CI) in all eight patients. These results strongly suggest that Lin-/CD34+ HPCs in bone marrow are not a source of persistent HIV-1 in patients on long-term suppressive therapy. The striking lack of HIV genetic evolution in the HIV populations after years of therapy strongly indicates little or no viral replication in the majority of cells from peripheral blood and GALT during suppressive therapy. The finding of multiple T cells with identical replication incompetent virus after long-term therapy is strong evidence that this persistent virus was due to expansion of cells with integrated proviral DNA rather than active viral replication.
In summary, these findings were important in several ways:
1. The second study to refute the Collins Nature Medicine papers, and find no evidence of HIV in carefully purified Lin-/CD34+ HPCs. Parallel data from the Siliciano lab has recently been reported at meetings.
2. Another careful study to find a lack of viral evolution in the presence of long-term suppressive ART. Again consistent with the complete interruption of effective rounds of replication by ART, like that of Kearney and the NCI Frederick group presented at a prior CROI meeting. This again highlights that the measurements of HIV DNA will in some part be measuring viral genomes that are functionally dead, and that these HIV DNA "fossils" may even expand if they are lodged in CD4 cell clones that are expanding.
Tae-Wook Chun (NIH, abstr. 13) reviewed potential mechanisms of HIV persistence, prospects for eradication, and new therapeutic approaches. Of note, he remarked that studies in his laboratory had found that histone deacetylase (HDAC) inhibitors induce HIV RNA expression from the cells of patients, but at levels much lower than seen with full mitogen activation. In such a setting, he also noted, exposure to HDAC inhibitors and HIV RNA production did not seem to result in cell death. Javier Martinez-Picado (Barcelona, abstr. 14) reviewed data from his widely-cited Nature Medicine paper (Buzon et al.) in which a minority of patients (13 or 48) who underwent raltegravir intensification were found to have an increased frequency of detectable HIV circular DNA forms. The genetic structure of these episomes differed from that of integrated proviral HIV-1 DNA forms, leading to the hypothesis that these viruses originated from different anatomical compartments. He presented a model that hypothesized the presence of a reservoir that intermittently released of infectious virus, leading to limited numbers of de novo infection events. Although Martinez-Picado agreed that there was no definitive evidence that more aggressive antiretroviral treatment strategies might reduce these intermittent rounds of new infection, he proposed that ART intensification would be needed when coupled with HIV-1 transcriptional inducers and immune-therapies to achieve cure.
Una O'Doherty (UPenn, abstr. 25) reported findings using a PCR technique developed in her laboratory to precisely and sensitively measure integrated HIV DNA. She found that integrated HIV DNA is present at very low levels in elite suppressors (HLA B57 or 27+) while 2-LTR circular forms are present at high levels. Intermediate levels were found in patients on ART, and the highest levels in untreated patients. However, the highest level of 2LTR circular, episomal HIV DNA was found in elite suppressors, consistent with the reports of others that there is a selective natural block to HIV integration in such patients.
Like the prior 2 speakers, Nicolas Chomont and Claire Vandergeeten (Vaccine and Gene Therapy Institute of Florida, abstr. 27 and 29) discussed the "persistent reservoir" of HIV infection from the point of view of HIV DNA. 95% of HIV DNA in these studies is found in CD4+ T cells, even prior to ART. ART substantially reduced the number of cells carrying proviral DNA in both blood and tissues. On average, he finds 300 integrant copies of HIV DNA per million T cells (range 10-2000). The size of this DNA reservoir is predicted by the CD4/CD8 ratio, as well as CD4 nadir and the CD4 increase on ART (r = 0.56 and 0.52). Duration of viremia prior to ART and age are also correlated, although more weakly. Only CD4 nadir is correlated in multivariate analysis. Overall, this suggests that the DNA measure is merely a surrogate for exposure to viremia and progression of disease over time. Chomont then also discussed previously published data showing a predominance of persistent infection as measured by DNA in central and transitional memory cells, and the role of IL7 and IL15 in inducing non-specific proliferation of CD4 cells. When such proliferation occurs, due to homeostatic expansion driven to maintain CD4 cell balance, HIV DNA integrants, some replication-competent and some not, may be able to be replicated along with the cell that carries them, without viral expression and resultant cell clearance. Therefore one strategy to reduce the pool of latently infected cells might be to interfere with the mechanisms responsible for the homeostasis of the memory T cell compartment. It remains to be seen if this strategy of "killing T cell memory to save T cells" will succeed. In ACTG 5214, 10 patients on ART received IL7. An increase in Ki67, PD-1, and HLA-DR were seen, followed by a decline over 4 wks. In 6 pts assayed, an increase of integrated HIV DNA was seen in 2 patients with CD4 cell counts of nearly 400, but not in those with counts below 400.
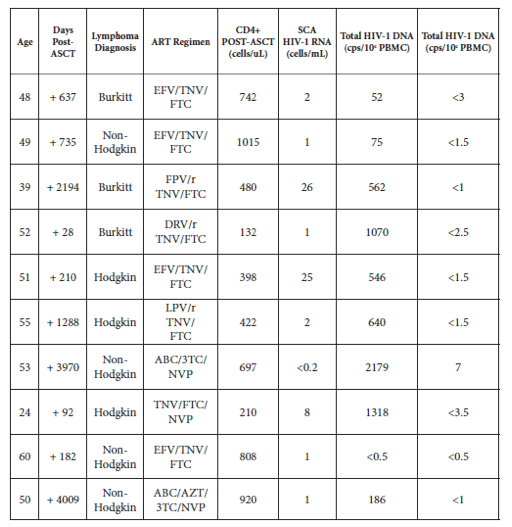
The only reported case thought to represent a cure of HIV infection, the "Berlin patient," underwent an allogeneic hematopoietic stem cell transplantation (ASCT) with stem cells that were naturally resistant to HIV infection as they were derived from a CCR5 delta 32 homozygous donor. John Mellors (U Pitt, LB abstr. 2) sought to understand the contribution of HSCT itself to this result by the study of HIV+ patients who underwent intensive myeloablative chemotherapy and ASCT for malignancies. 10 patients were studied. Clinical characteristics, HIV-1 RNA from plasma, total HIV-1 DNA and 2-LTR circles from PBMC post-ASCT are shown below. Persistent viremia was detected in 9 of 10 patients post-SCT, with a median HIV-1 RNA of 1.5 copies/mL (range: <0.2 to 26). Similarly, HIV-1 DNA was detectable post-ASCT in 9 of 10 patients, with a median total HIV-1 DNA of 554 copies/106 PBMC (range: <0.5 to 2179). 2-LTR circles were detectable post-ASCT in only 1 of 10 patients. Notably, the only patient with undetectable plasma HIV-1 RNA had the highest levels of HIV-1 DNA and detectable 2-LTR circles. Additionally, plasma viremia was detectable in the patient with undetectable HIV-1 DNA in PBMC. Mellors noted that of the 5 x 1010 cells infused and only 5 x 108 were CD34+ cells, and that many of the transplanted cells might be resting CD4 T cells, cells that would be somewhat resistant to the killing effect of chemotherapy due to their resting state.
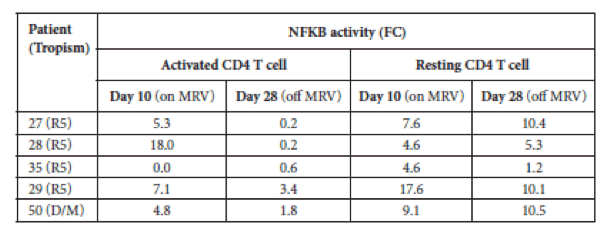
An unusual but potentially interesting finding was reported by Moreno's group from Madrid (LB abstr. 4), who suggested that maraviroc (MVC) can activate NFkB in patients regardless of the tropism of their infecting virus. Like the Buzon study that found that RAL intensification increased 2LTRs circular HIV DNA forms, this group recently found that circles also increased in patients who received ART intensification with MVC (Gutierrez C, et al. PLoS ONE, 2011). The group then sought to test if MVC could increase HIV-1 transcription (although how that would lead to an increase in circular DNA forms is unclear to this writer), and measured NFkB and the related protein NFAT in the nuclei of activated and resting CD4 cells before and after 10 days of MVC monotherapy, and 18 days after MVC withdrawal. There was a quantitative change in the level of the NFkB p65 protein in the nuclei of both resting and activated cells in most patients, which usually declined after MVC withdrawal. As the patients were on no other ART, this could be directly due to MVC effect on cells, and obviously most of the cells assayed here in the patients are not infected. It was not mentioned what effect on viral load was seen in the first 10 days of MVC monotherapy. And as a comparative control, there was no change in NFAT. The authors suggested that MVC might therefore have some latency-purging effects, in addition to its designed antiviral ones, but this requires much more study to confirm this hypothesis.
Puertas and Massanella (IrsiCaixa - AIDS Research Institute, Barcelona; abstr. 41 & 42) reported that MVC intensification of ART sped on the decay of HIV-1 DNA in PBMC of recently infected patients treated with TDF/FTC and RAL. The results of the study might be seen as consistent with but not proof of the above hypothesis. A total of 30 recently (<24 weeks) HIV-1-infected patients with CCR5-using viruses were randomly assigned to initiate a maraviroc intensified triple-drug therapy (n=15) or a standard HAART (n=15). Blood samples were collected and PBMC stored at study entry (BL) and weeks 2, 4, 12, 24 and 48. From every sample, genomic and episomal DNA was isolated separately. From the genomic fraction, total HIV-1 DNA was quantified by qPCR using internal LTR primers, and integrated HIV-1 DNA was measured by using the LTR-Alu system. Unintegrated HIV-1 DNA, from the episomal DNA fraction, was evaluated by qPCR using specific primers for 2-LTR circles. Total HIV-1 DNA in PBMC was progressively and significantly reduced, from baseline to week 48, in both treatment groups. Although there were no differences at the end of the study, at week 24 the difference between groups was statistically significant (p=0.034), suggesting a relative delay in the decay of the total viral reservoir in the control group. When analyzing integrated HIV-1 DNA, similar reduction dynamics were observed in both the intensified and the control groups. Nevertheless, the reduction from BL to week 48 was only significant in patients receiving maraviroc. A rapid and significant decrease of PD-1 in total or CD45RO+CD8+ T-cells was observed compared to baseline in both groups (P<0.01 in all cases). In general we observed a trend towards a slower decrease in activation markers in the MVC arm, which could be a consequence of the high CCR5 expression in activated CD8 T-cells. Overall, no major differences in latent reservoir size were observed at 48 weeks between both groups, although maraviroc intensification of a triple-drug therapy results in a faster control of viral replication.
Martin Markowitz (Aaron Diamond AIDS Research Center, abstr. 31) reported the results of his single-site study of 3- vs. 5-drug therapy in early/acute HIV infection (mean 50 days after estimated date of infection). He compared TDF/FTC and ATZ/r or DRV/r) to therapy with those agents and RAL and MVC. 23 of 26 patient completed 96 weeks of 5-drug therapy, compared with 11 of 14 on standard therapy. As expected the 5-drug regimen achieved <50 copies/ml of plasma viremia faster, but at week 48 there were 3 virological failures in the 5 drug arm, and none in the 3-drug arm. Decay of cell-associated RNA and proviral DNA decay was identical. CD4 cell counts in blood and GALT are not different between arms, and levels of immune activation and proliferation measured by standard markers on CD4 and CD8 cells were no different, and quite similar to uninfected patients. The size of the latent reservoir was determined as per Chun et al. 1997, and notably the size of the latent reservoir did not correlate with baseline HIV-1 RNA, duration of infection prior to treatment initiation, levels of plasma viremia at 48 weeks or detectability at week 48 using a 1-copy assay, or levels of proviral DNA or cell associated RNA at week 48. The size of the latent reservoir after 48 weeks of treatment correlated with levels of proviral DNA and cell associated RNA pretreatment and was inversely correlated with baseline % CD4. Finally, there were no statistically significant differences in the % of CD8+HLA+DR+ T cells at week 48 and levels of sCD14 in plasma at week 48 and 96 in suppressed HIV+ subjects treated with either 3- or 5-drug ART when compared to a group of 13 normal volunteers, suggesting that the early initiation of therapy may prevent residual immune activation that often accompanies treatment initiated later in the course of therapy. Overall, although there appeared to be some potential benefit of intensive early therapy, the effects appeared small.
Without reiterating all the details, Ananworanich (Thai Red Cross AIDS Research Center, Bangkok, Abstr. 32) reported a nearly identical study performed in Thailand. The notable difference here was simply the impressive number of patients treated in truly acute infection --- all treated within 3-4 weeks of infection. Again, although some marginal differences were seen, overall there was no advantage of 5-drug over 3-drug therapy. Unlike the NYC study, this group did not see more virological failures in the 5-drug arm.
Buzon (abstr. 33) now at MGH rather than Madrid, reported that there was a reduction in HIV-1 reservoir size after 10 years of ART started in primary infection. The "reservoir" here measured was integrated and total HIV DNA, more than 10 years of ART reducing integrated and total HIV-1 DNA to levels observed in elite controllers. Although the authors concluded that this finding suggested that prolonged ART initiated during primary infection may allow for a clinically significant depletion of HIV-1 reservoirs, this seemed like a long time to wait for this effect.
Janice Clements (Johns Hopkins, abst 36) outlined the characteristics of infection and persistence seen in their model of accelerated SIV CNS infection in the pigtailed macaque (Macaca nemestrina). When treated with a four-drug antiretroviral regimen, this SIV model mimics HIV infection in humans on ART including suppression of plasma viremia and rebound of CD4+ T cells. During acute infection both CD4+ lymphocytes as well as monocytes and macrophages are infected. Clements showed that the spleens of SIV infected animals contain significant numbers of infected macrophages.
SIV-infected, CD68+ macrophages are detectable in spleen at 10 days post-infection and the number of these cells increases by terminal infection in untreated, infected pigtailed macaques. SIV DNA and RNA are detectable in the spleen of HAART treated animals at terminal infection, at about 50 copies/μg total RNA.
Sharon Lewin (Burnett Inst., Melbourne, abstr. 38) reviewed the establishment of latency in her published primary cell model, in which latency is established by direct infection of resting CD4+ T-cells following incubation with chemokines that bind to the chemokine receptors CCR7 (CCL19 and CCL21), CXCR3 (CXCL10) and CCR6 (CCL20). HIV integration in resting CD4+ T-cells was said to be dependent upon activation of the PI3 kinase pathway. Myeloid dendritic cells, were also said to increase latent infection of memory but not naïve CD4 cells. Finally gene array analysis of latently infected CD4+ T-cells, derived following co-culture with DCs, showed significant differential expression of 428 genes including induction of genes known to arrest or delay cell cycle progression, such as ATF3 and GADD45A, IRF7, IL15, DDIT3, and ATF3; suppression of multiple genes required for active cell cycle, including CDC20 and CDK1; and suppression of genes required for NF-κB activation.
Gene therapy approaches were discussed in one in-depth session. Paula Cannon (USC, abstr, 44) reviewed her work in the humanized mouse model, attempting to optimize gene modification within hematopoietic stem and progenitor cells (HSPC) to disrupt the CCR5 gene while minimizing any adverse effects on cell viability or hematopoietic potential. Autologous stem cells were engineered to be CCR5-negative using designer nucleases. The enzymes are designed with a nuclease domain and DNA-binding domain, utilizing the host non-homologous end-joining pathway to repair damage with focal mutations that disrupt the function of the target gene (herein CCR5). Two kinds of enzymes, that target either 3-base-pair or 4-basepair sequences have a cutting efficiency of 30% in cell lines, but as little as 4-17% in HSPC. Goals are to scale-up to adult HSC targets, alter cells in sufficient numbers to dose a human, achieve >5% CCR5 disrupt, >80% viability and retention of stem cell properties, prior to testing in patients. Eventually, the first studies will be piloted in AIDS lymphoma patients, likely using nucleases transduced with adenoviral vectors into T (not stem) cells. Much work remains to be done.
Carl June (U Penn, abstr. 45) reported the results as seen at ICAAC of a single dose of zinc finger nuclease (ZFN) CCR5 modified autologous CD4 T cells to patients. June concluded that this vector infusion increases CD4 counts that persist over time. CCR5 modified CD4 cells expand rapidly and home to the gut. June also pointed out that an allogeneic HSPC graft contains 106 CD34+ stem cells, but also 3 x 108 contaminating T cells (ie 30-fold more). Therefore the Berlin patient received a lot of CCR5-negative CD4 cells, as well as stem cells, at transplant. As reported at ICAAC, CD4 counts rose as much as 1800 cells after transplant, then declined by 90 days to about 200-300 cells over baseline. The increase was all CD4 cells. qPCR confirmed that the expected CCR5 pentamer duplications were seen and were stable, raising the question of whether the cells were long-lived because they were senescent and had left the cell cycle. During a planned ART interruption in 6 patients, viral load rebounds reached to nearly setpoint, and seemed to start to decline around 70 days prior to restart. June concluded that the reinfusion of 10-30 billion cells is safe, that 1.2-30% of blood CD4s were edited, and that cells traffic to gut and persist there.
Jan van Lunzen (Hamburg, abstr. 47) reviewed the original gene therapy approaches piloted in Germany, and current approaches to excise HIV proviral DNA. This work has been published (van Lunzen Mol Ther 2007; Sarkar Science 2007; Bucholz & Hauber 2011 Methods. The group now seeks to use a self-inactivating retroviral vector to alter gene in CD34 stem cells, and reimplant these cells into a humanized mouse model.
Finally Keith Jerome (Fred Hutchinson Cancer Research Center, abstr. 48) discussed studies of the use of novel enzymes known as homing endonucleases (HEs), which specifically target long (~22 base pair) sequences in DNA and induce double strand breaks. These breaks are subsequently repaired by error-prone non-homologous end joining, thus allowing targeted disruption of essential viral genes.
These elegant tools can be designed to target a variety of sites within the HIV genome, including Env, Pol, Tat, and Vif, but the specific and safe delivery of these large molecules to the rare latent genomes within an infected person is a very significant hurdle to be overcome before clinical use can be tested.
Perhaps the most interesting presentation (abstr. 35) of the meeting was a trio of talks by Timothy Schacker (U Minnesota), Courtney Fletcher (U Nebraska), and Mario Stevenson (U Miami), analyzing virions by in situ hybridization, intracellular drug levels, and HIV DNA forms in the lymph node tissue of patients on ART. 12 patients, mean age 33, 5 yrs HIV+, nadir CD4 ca. 350 cells, were intensively studied for the first 12 months of ART administration. But limited data was so far available. All were treated with TDF and FTC, and four with atazanavir, one with darunavir, and 4 with efavirenz. Excellent adherence was reported, and corroborated by plasma drug levels. Suppression was achieved in all patients, although details were scant, and no virologic failures were reported. Biopsies of GALT and inguinal nodes were done, as was intensive PK at month 1, 3, and 6 of therapy. Mass-spectrometer measurements of TDF-DP; FTC-TP; ATV; DRV; EFV; RAL down to levels of 2.5 fmol/million cells were performed in blood and tissue. TFV-DP concentrations in blood averaged 60 fmol as expected, and tissue levels appeared generally similar if not sometimes higher. However tissue levels of FTC-TP were generally lower than the target level seen in plasma. ATV blood levels were generally as expected, and EFV blood levels were as expected but some tissues lower! ATV levels were low in some tissues, and EFV tissue levels were quite unpredictable but often low. So, specific differences were seen for different drugs in different compartments, even within the same drug class.
But controls (parent drug in tissue, spiked samples) were not shown, and the PK data was from an N = 4. Nevertheless, the results were dramatic. Does this mean that levels of many drugs are inadequate in the tissue? Can these results be confirmed and extended? Are levels of active drug-DPs and drug-TPs in tissue that are thought to be inadequate (as they are lower than plasma levels) really sufficient to suppress replication in tissues? Is viral expression really more defective than we suspect, or innate host defenses (eg ApoBEC) really doing a better job, such that even weak drug pressure is sufficient? What does it mean that levels are so low in these patients in whom therapy is succeeding, and drug-resistance is not emerging?
In situ studies of HIV RNA in lymphoid tissue were equally striking. Abundant but widely distributed virion particles were seen in one lymphoid follicle, but an adjacent one seemed completely devoid of virus. Although the presence of these viral particles suggested the local expression and retention of virions in follicles of lymph tissue, the absence of particles in an adjacent tissue in a patient who had untreated full-blown viremia only a few months before suggested that such particles must be cleared over a few months. Indeed, a presenter said that 3 of 4 patients had decay of HIV RNA (or at least fewer in situ grains seen) seen in serial samples on ART. The presenters proposed that their observations comprised evidence of intermittent, ongoing, single-round infections without ongoing spread of infection ---- creating a "reservoir" that was persistent but dynamic and not quiescent. Clearly more work needs to be done to understand what is going on, and to clearly interpret the meaning of these "snapshots" of viral RNA that have been taken. One thing patients should not do is take higher doses of ART than is recommended.
| |
| |
| |
|
|
|