| |
Faldaprevir combined with pegylated interferon alfa-2a and ribavirin in treatment-naïve patients with chronic genotype1 HCV: SILEN-C1 trial
|
| |
| |
Download the PDF here
HEPATOLOGY, June 2013
Mark S. Sulkowski,1 Tarik Asselah,2 Jacob Lalezari,3 Peter Ferenci,4 Hugo Fainboim,5 Barbara Leggett,6 Fernando Bessone,7 Stefan Mauss,8 Jeong Heo,9 Yakov Datsenko,10 Jerry O. Stern,11 George Kukolj,12Joseph Scherer,11 Gerhard Nehmiz,10 Gerhard G. Steinmann,10 and Wulf O. Bocher10
In this large, phase 2 study of faldaprevir QD, in combination with PegIFN/RBV, cure of infection (SVR) was achieved in up to 84% of HCV GT-1 patients, with more than 80% meeting VR criteria for shortened treatment duration (24 weeks). Overall, the treatment regimen was safe and tolerable. Confirmatory phase 3 trials testing 120 and 240 mg QD faldaprevir without LI, in combination with PegIFN/RBV, are ongoing in treatment-naïve and -experienced patients, as well as patients with HCV/HIV coinfection.
Abstract
Faldaprevir (BI 201335) is a potent, hepatitis C virus (HCV) NS3/4A protease inhibitor with pharmacokinetic properties supportive of once-daily (QD) dosing. Four hundred and twenty-nine HCV genotype (GT)-1 treatment-naïve patients without cirrhosis were randomized 1:1:2:2 to receive 24 weeks of pegylated interferon alfa-2a and ribavirin (PegIFN/RBV) in combination with placebo, faldaprevir 120 mg QD with 3 days of PegIFN/RBV lead-in (LI), 240 mg QD with LI, or 240 mg QD without LI, followed by an additional 24 weeks of PegIFN/RBV. Patients in the 240 mg QD groups achieving maintained rapid virologic response (mRVR; viral load [VL] <25 IU/mL at week 4 and undetectable at weeks 8-20) were rerandomized to cease all treatment at week 24 or continue receiving PegIFN/RBV up to week 48. VL was measured by Roche TaqMan. Sustained virologic response (SVR) rates were 56%, 72%, 72%, and 84% in the placebo, faldaprevir 120 mg QD/LI, 240 mg QD/LI, and 240 mg QD groups. Ninety-two percent of mRVR patients treated with faldaprevir 240 mg QD achieved SVR, irrespective of PegIFN/RBV treatment duration. Eighty-two percent of GT-1a patients who received faldaprevir 240 mg QD achieved SVR versus 47% with placebo. Mild gastrointestinal disorders, jaundice resulting from isolated unconjugated hyperbilirubinemia, and rash or photosensitivity were more common in the active groups than with placebo. Discontinuations resulting from adverse events occurred in 4%, 11%, and 5% of patients treated with 120 mg QD/LI, 240 mg QD/LI, and 240 mg QD of faldaprevir versus 1% with placebo. Conclusion: Faldaprevir QD with PegIFN/RBV achieved consistently high SVR rates with acceptable tolerability and safety at all dose levels. The 120 and 240 mg QD doses are currently undergoing phase 3 evaluation.
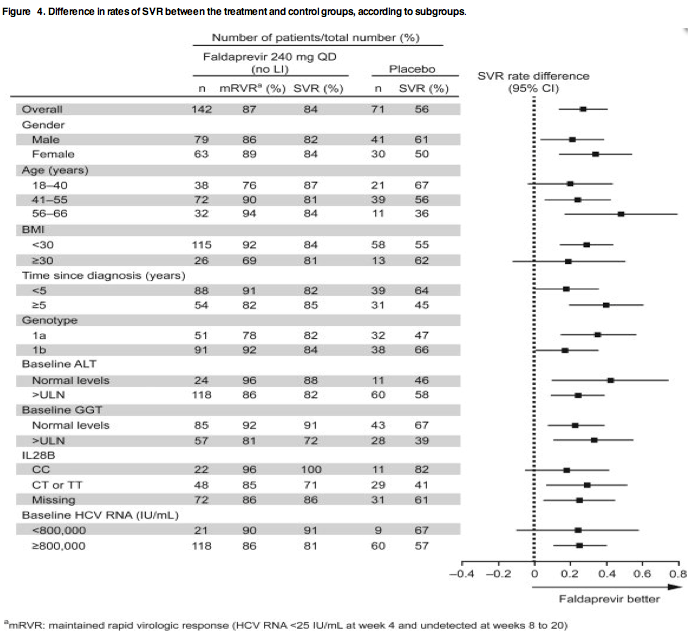
Figure 4. Difference in rates of SVR between the treatment and control groups, according to subgroups.
Treatment of hepatitis C infection has advanced since its initial characterization.1 Hepatitis C virus (HCV) genotype (GT)-1 represents the most common GT in many parts of the world and, historically, has been less responsive to peginterferon alfa-2A (PegIFN) and ribavirin (RBV), compared with other HCV GTs, despite longer treatment duration (48 weeks) and higher-dose RBV. Since the proof-of-concept study with the HCV NS3/4A peptidomimetic protease inhibitor (PI), BILN 2061,2 multiple PIs have entered clinical development. Two PIs, the α-ketoamide derivatives boceprevir and telaprevir, have been approved for treatment in many regions of the world. Taken three times daily in combination with PegIFN/RBV, both boceprevir and telaprevir have significantly improved sustained virologic response (SVR) rates and shortened treatment duration in approximately half of treated HCV GT-1 patients, as compared with PegIFN/RBV alone.3-6 However, both agents carry a high pill burden and add significant side effects to those of PegIFN/RBV, including severe skin rashes/pruritus (telaprevir), anal discomfort (telaprevir), dysgeusia (boceprevir), and anemia (telaprevir and boceprevir).
Faldaprevir is a potent, once-daily (QD), HCV NS3/4A PI7 with antiviral activity in in vitro HCV subgenomic replicon assays, as well as NS3 protease assays derived from HCV GT-1, -4, -5, and -6.8 Preclinical and human pharmacokinetic (PK) studies suggested that faldaprevir has a long half-life, consistent with QD dosing.9 Phase 1b studies demonstrated that faldaprevir QD combined with PegIFN/RBV was well tolerated and induced strong antiviral responses in treatment-naïve and -experienced HCV GT-1 patients.10 Here, we report on the results of a phase 2b, multicenter, randomized, double-blind study of faldaprevir or placebo in combination with PegIFN/RBV in treatment-naïve, HCV GT-1-infected patients (SILEN-C1; Safety, and antIviraL Effect of faldaprevir iN hepatitis C).
Abbreviations
AE, adverse event; ALT, alanine aminotransferase; BMI, body mass index; CI, confidence interval; DRESS, drug rash with eosinophilia and systemic symptoms; EVR, early virologic response; GGT, gamma-glutamyl transferase; GT, genotype; HCV, hepatitis C virus; HIV, human immunodeficiency virus; IVRS, interactive voice response system; LI, lead-in; LLOD, lower limit of detection; LLOQ, lower limit of quantification; mRVR, maintained rapid virologic response; OR, odds ratio; PCR, polymerase chain reaction; PegIFN, pegylated interferon alfa-2a; PI, protease inhibitor; PK, pharmacokinetic; PR, peginterferon/ribavirin; QD, once-daily; RBV, ribavirin; RGT, response-guided therapy; SJS, Stevens-Johnson syndrome; SVR, sustained virologic response; UGT, uridine diphosphate glucuronosyltransferase; ULN, upper limit of normal; VL, viral load; VR, virologic response.
Results
Patient Disposition and Baseline Characteristics.
Of 581 patients screened, 429 were randomized to treatment, whereas 152 did not meet entry criteria (Fig. 2). Of the 429 treated patients, 74 prematurely discontinued the trial medication (faldaprevir or placebo). Reasons for treatment discontinuation included AEs (n = 28), lack of efficacy (n = 23), refusal to continue the trial medication (n = 8), noncompliance with the protocol (n = 5), loss to follow-up (n = 3), and other reasons (n = 7).
Patients were evenly distributed over all dose groups with respect to gender, race, HCV RNA, GT, age, BMI, and IL28B GT (Table 1). Patients were predominantly male (55%), mean age was 46 ± 11 years, mean BMI was 26.0 ± 4.6, and mean HCV RNA was 6.39 ± 0.61 log10 IU/mL. Analysis of IL28B GT (rs12979860) was performed retrospectively in 223 patients (the remaining patients did not provide consent for testing); of those tested, 27% were CC and 73% were non-CC. Six patients enrolled into the study and received study drug based on initial testing, but were found not to be infected with GT-1 by sequencing (GT-3, n = 2; GT-4, n = 1; GT-6, n = 3); all non-GT-1 patients achieved SVR. These patients are included in the safety analysis, but were excluded from the efficacy analysis.
Efficacy.
SVR was achieved by 56% of patients in the placebo arm, compared to 72% of patients in the 120 mg QD/LI arm (P = 0.054), 72% of patients in the 240 mg QD/LI arm (P = 0.021), and 84% of patients in the 240 mg QD arm (P = 0.001; Fig. 3). The majority of patients treated with faldaprevir achieved mRVR with rates of 15%, 80%, 78%, and 87% in the placebo, 120 mg QD/LI, 240 mg QD/LI, and 240 mg QD dose groups, respectively (Fig. 3). Virologic response (VR) is summarized in Table 2. In the placebo group, 11 of 71 patients (15%) relapsed. Thirty-one of 352 patients (9%) treated with faldaprevir relapsed: five of 69 (7%) in the 120 mg QD/LI group, 15 of 141 (11%) in the 240 mg QD/LI group, and 11 of 142 (8%) in the 240 mg QD group.
In the 240 mg QD/LI and 240 mg QD dose groups, 78% and 87% of patients, respectively, achieved mRVR and were eligible for rerandomization to either 24 or 48 weeks of PegIFN/RBV (Fig. 3). High rates of SVR (81% to 96%) were observed in patients with mRVR. In the 240 mg QD/LI arm, lower SVR (81% versus 96%; P = 0.051) and higher relapse (17% versus 4%) rates were observed in patients who achieved mRVR and were treated for 24 weeks, compared with those treated for 48 weeks. In contrast, 107 of 116 patients (92%) in the 240 mg QD dose group achieved SVR, irrespective of the duration of treatment (24 versus 48 weeks; P = 1.000); rates of relapse were also similar (5% versus 2%).
In patients who received the 240 mg QD dose without LI, SVR rates were consistently high across a wide range of patient subgroups (Fig. 4). Notably, the SVR rate for GT-1a patients was 82% (compared with 47% on PegIFN/RBV; P = 0.0013). All 22 patients (100%) with IL28B GT CC achieved SVR, compared with 82% with PegIFN/RBV alone. In this arm, the percentage of patients achieving mRVR and thus eligible for 24 weeks overall treatment duration was 87%, with similar rates across all subgroups, including non-CC patients. A univariate logistic regression analysis of baseline predictors of SVR in patients in the 240 mg QD dose group, considering IL28B GT, ALT, GGT, HCV VL, gender, BMI, and time since diagnosis, indicated that only GGT (P = 0.013, odds ratio [OR; 95% CI] = 3.32 [1.29, 8.59]) and IL28B (P = 0.154) achieved the predefined significance level for multivariate testing. However, because 100% of patients with the IL28B CC GT achieved SVR, multiple variable regression was not performed. Within the 48 non-CC GT patients, the effect of GGT was slightly reduced (OR [95% CI] = 2.03 [0.56, 7.3]).
Rates of virologic breakthrough during faldaprevir treatment were comparable (3%-6%) with that observed in the placebo group (3%), but, in contrast to the placebo group, were predominantly associated with the selection of NS3 R155K or D168V variants for GT-1a or GT-1b patients, respectively. Three patients (1%) in the active arms experienced virologic rebound during PegIFN/RBV therapy after stopping faldaprevir therapy, including one R155K change (GT-1a), one without detectable resistant mutant (GT-1a), and one (GT-1a) with mixed substitutions at R155 and D168. Post-treatment rebound in the active arms from the nonresponder group was characterized by two R155K substitutions (one GT-1a in each 240 mg dose group) and the lack of detectable resistant mutants in the other five patients. Relapses among the faldaprevir treatment groups were observed in 31 of 352 patients (9%) with an undetectable VL at the end of treatment; viral sequencing of this patient group was characterized by predominant selection of R155K variants in GT-1a patients (eight of 12) and D168V variants in GT-1b patients (11 of 19). Notably, five of six patients in the faldaprevir treatment groups with resistance substitutions detectable before treatment (two R155K in GT-1a and four D168 in GT-1b) achieved SVR.
Safety.
Most AEs observed during or up to 30 days after the end of treatment with faldaprevir were those commonly related to PegIFN/RBV therapy (Table 3). Serious AEs were reported in two patients (3%) in the placebo group and 33 (9%) treated with faldaprevir (all serious AEs are described in the footnote of Table 3). Discontinuation of treatment resulting from an AE was lower for patients treated with placebo versus faldaprevir: one of 71 (1%) discontinued treatment in the control group, compared to three of 69 (4%), 16 of 140 (11%), and eight of 149 (5%) patients treated with 120 mg QD/LI, 240 mg QD/LI, and 240 mg QD faldaprevir (Table 3). Of the 28 patients who discontinued faldaprevir or placebo because of AEs, 25 also discontinued PegIFN/RBV.
The primary AEs attributed to faldaprevir were mild gastrointestinal disorders (nausea, diarrhea, and vomiting), jaundice, pruritus, and rash. All AEs resolved during or after end of treatment. Rash and photosensitivity were higher in the 240 mg groups, compared to placebo and the 120 mg group. Probability of occurrence of rash or photosensitivity reactions tended to increase with increasing median trough plasma levels of faldaprevir. Rashes had an erythematous, macular, or papular morphology, preferentially affecting the trunk, arms, and legs, and usually occurred during the first 12 weeks of treatment. Mucus membranes and other organs were not affected in any patient. There were no cases of Stevens-Johnson syndrome (SJS), erythema multiforme, or drug rash with eosinophilia and systemic symptoms (DRESS). The majority of cases were mild and were managed without treatment interruption, in most cases, by applying topical treatments and, rarely, systemic corticosteroids (<5%). Photosensitivity mostly manifested as mild erythema limited to sun-exposed areas of the body and led to one discontinuation. Severe rash events were observed in 11 patients (3% in the faldaprevir groups): five patients (4%) in the 240 mg QD/LI group and six (4%) in the 240 mg QD group. Ten patients in the 240 mg dose groups (3%) discontinued faldaprevir because of rash, which was classified as severe in eight of these cases.
At the higher doses of 240 mg QD, 20% to 25% of patients experienced jaundice (usually mild) as a result of dose-dependent, isolated, unconjugated hyperbilirubinemia associated with faldaprevir; rates of mild jaundice were similar for placebo and 120 mg QD (Table 3). Hyperbilirubinemia was rapidly reversible (bilirubin normalized in all patients subsequent to cessation of faldaprevir dosing) and not associated with liver injury, hemolysis, or any other clinical symptoms. Changes in safety laboratory values were consistent with those observed with PegIFN/RBV (Table 4). Importantly, there was no additional effect of faldaprevir on hemoglobin levels or white blood cells, compared to the control group; the rate of erythropoietin was similar in the placebo and active arms (5% to 12%).
Discussion
The PI faldaprevir, dosed QD in combination with PegIFN and RBV, increased SVR rates of treatment-naïve GT-1 patients from 56% in the placebo group to as high as 84% with 240 mg faldaprevir. SVR rates were similar with this dose in patients with historically less-favorable IL82B GT and HCV GT-1a. Interestingly, patients in the 120 mg QD/LI group had similar EVR and SVR rates to those in the 240 mg QD/LI group, suggesting a lack of a dose effect on VR at the higher dose. Furthermore, although not directly compared, SVR rates observed with all dose regimens of faldaprevir and PegIFN/RBV appear at least similar to those reported with the recently approved PIs boceprevir and telaprevir (68% and 73%, respectively).3, 5
Response-guided therapy (RGT) was assessed in both 240 mg dose groups, where 78% and 87% of patients with or without LI achieved mRVR, respectively, defined as HCV RNA < LLOQ at week 4 and < LLOD at weeks 8 to 20. Thus, the majority of patients met the response criteria for rerandomization to either stopping all treatment after 24 weeks or continuation of PegIFN/RBV to week 48. All patients who were not eligible for rerandomization continued PegIFN/RBV to week 48. Importantly, patients achieving mRVR who stopped all treatment at week 24 in the 240 mg QD dose group showed equivalent SVR rates as those who continued PegIFN/RBV to week 48. Because these data were derived in a randomized fashion, they support the application of RGT with 24 weeks of overall treatment duration for treatment-naïve GT-1 patients achieving mRVR.
Clinical trials of boceprevir and telaprevir also included the option for shortened treatment duration, but incorporated more-stringent criteria (which included HCV RNA < LLOD at week 4) than this study.3, 5 When the criteria used in the telaprevir studies to identify patients eligible for shortened treatment duration (HCV RNA < LLOD at weeks 4 and 12) were applied to this data set, up to 75% met the criteria, suggesting that the majority of patients treated with faldaprevir plus PegIFN/RBV may be eligible for 24 weeks of therapy. Furthermore, these data support the application of less-stringent criteria at treatment week 4 (<25 IU/mL detected or undetected), which may be acceptable for the determination of patients who can be treated for shorter duration with faldaprevir plus PegIFN/RBV.
HCV NS3/4A PIs have been shown to rapidly select for the emergence of resistance mutations when administered as monotherapy.10, 12 Therefore, the effect of 3 days of pretreatment of patients with PegIFN and RBV before adding faldaprevir was assessed directly by comparing 240 mg QD dose groups with or without LI. The rationale was that the short delay of the first intake of faldaprevir would prevent the possibility of functional PI monotherapy. Surprisingly, the administration of a 3-day LI with PegIFN/RBV before initiation of faldaprevir resulted in approximately 10% lower rates of mRVR and SVR, compared with faldaprevir initiated simultaneously with PegIFN/RBV. A similar negative effect was observed in a second independent phase 2 trial of faldaprevir in patients who had nonresponse to previous PegIFN/RBV treatment.13 Although the reasons for poor response with short LI are unknown, rapid and profound inhibition of HCV replication might restore IFN responsiveness, as suggested by the decrease of plasma levels of IFN-inducible protein 10, a lymphocyte chemokine indicative of endogeneous activation of host IFN pathways, in HCV patients treated with IFN-free regimens.14 However, further research is required to test this hypothesis. Although the reasons for lower response rates with 3-day LI are not fully understood, this strategy was not selected for further investigation.
Breakthroughs and relapses in the faldaprevir treatment groups were rare and usually associated with the selection of common NS3 PI-resistant variants, whereas patients failing PegIFN/RBV had wild-type NS3 sequences detected, with the exception of one patient who had selected for a D168E mutant, a previously observed natural polymorphism that also shifts potency of some PIs. Faldaprevir selected NS3 mutants predominantly encoded for R155K and D168V in GT-1a and GT-1b, respectively. Patients who experienced postfaldaprevir treatment rebound (either relapser and rebound from nonresponse) selected for similar NS3 amino acid changes, although 29% of isolates across the faldaprevir treatment groups encoded for virus that lacked known resistance mutants. In vitro studies have demonstrated 100-fold to 500-fold reduced sensitivity of various viral GT-1a strains carrying the R155K substitution, whereas D168V substitutions in GT-1b strains conferred >1,000-fold reduced sensitivity to faldaprevir.15 The clinical data indicate that these shifts in sensitivity cannot be offset by the increase in faldaprevir exposure achieved by dose doubling from 120 to 240 mg QD.
The most frequent AEs were those typical for PegIFN/RBV therapy. Although 120 mg QD faldaprevir had a safety profile similar to PegIFN/RBV (only pruritus and vomiting increased), higher frequencies were reported for skin rash, photosensitivity, jaundice, nausea, and diarrhea with the 240 mg QD dose. However, the vast majority of events were mild or moderate in intensity and only 4% to 11% of patients discontinued faldaprevir at 120 or 240 mg QD. Jaundice was, in all but one case, the result of isolated increases of unconjugated bilirubin (one patient with normal plasma bilirubin), was rapidly reversible after cessation of faldaprevir, and was not associated with signs of liver toxicity or excess hemolysis; patients had no other symptoms, and only one patient in the trial discontinued because of jaundice. In vitro studies demonstrated that faldaprevir mediated inhibition of the bilirubin-conjugating enzyme UGT1A1 and, to a lesser extent, the organic anion-transporting polypeptide 1 and multidrug-resistant protein 2 transporters, which appear to be the key drivers of this finding.16 This effect is comparable with that of other PIs in development for HCV17 or in clinical use for HIV (e.g., atazanavir) treatment, which is not considered a sign of hepato- or hematotoxicity.18 Skin rash and photosensitivity reactions, which were more frequent with the 240 mg QD dose, were usually mild and could be managed without treatment modifications, in most instances. There were few discontinuations resulting from skin events and no cases of DRESS syndrome, SJS, or erythema multiforme. Of note, there was no effect of faldaprevir on hemoglobin levels, red blood cell counts, or leukocyte counts, suggesting that anemia and leukocytopenia reported for some other PIs are not a class effect, but rather compound-specific side effects.
A potential limitation of this trial was the exclusion of patients with liver cirrhosis; this was because of the lack of phase 1 safety data in this patient population at the initiation of this study. However, tolerability, safety, and efficacy of 240 mg QD faldaprevir with PegIFN/RBV given for four weeks in patients with compensated liver cirrhosis (Child-Pugh score) was demonstrated to be similar to patients without cirrhosis in recently completed phase 1 and 2 trials.19, 20 Importantly, the PK characteristics were unchanged in patients with cirrhosis. In addition, degree of liver fibrosis was not collected prospectively in this study, precluding any analysis of the association of fibrosis stage on the efficacy and safety of faldaprevir. Lastly, because of the inclusion of RGT in some, but not all, treatment arms, the study was only fully blinded up to treatment week 24.
In this large, phase 2 study of faldaprevir QD, in combination with PegIFN/RBV, cure of infection (SVR) was achieved in up to 84% of HCV GT-1 patients, with more than 80% meeting VR criteria for shortened treatment duration (24 weeks). Overall, the treatment regimen was safe and tolerable. Confirmatory phase 3 trials testing 120 and 240 mg QD faldaprevir without LI, in combination with PegIFN/RBV, are ongoing in treatment-naïve and -experienced patients, as well as patients with HCV/HIV coinfection.
|
|
| |
| |
|
|
|