| |
Olysio (Simeprevir): FDA (preliminary prescribing information; summary of the basis of approval and highlights from the prescribing information
|
| |
| |
On November 22, 2013 FDA approved Olysio (simeprevir) 150 mg capsules for the treatment of chronic hepatitis C (CHC) infection, as a component of a combination antiviral treatment regimen. Below is a summary of the basis of approval and highlights from the prescribing information. Please refer to the full prescribing information for all the information needed to use OLYSIO safely and effectively.
INDICATIONS AND USAGE
OLYSIO is a hepatitis C virus (HCV) NS3/4A protease inhibitor indicated for the treatment of chronic hepatitis C (CHC) infection as a component of a combination antiviral treatment regimen.
OLYSIO efficacy has been established in combination with peginterferon alfa and ribavirin, in HCV genotype 1 infected subjects with compensated liver disease (including cirrhosis)
The following points should be considered when initiating OLYSIO for treatment of chronic hepatitis C infection:
OLYSIO must not be used as monotherapy
OLYSIO efficacy in combination with peginterferon alfa and ribavirin is influenced by baseline host and viral factors.
OLYSIO efficacy in combination with peginterferon alfa and ribavirin is substantially reduced in patients infected with HCV genotype 1a with an NS3 Q80K polymorphism at baseline compared to patients infected with hepatitis C virus (HCV) genotype 1a without the Q80K polymorphism. Screening patients with HCV genotype 1a infection for the presence of virus with the NS3 Q80K polymorphism at baseline is strongly recommended. Alternative therapy should be considered for patients infected with HCV genotype 1a containing the Q80K polymorphism.
OLYSIO efficacy has not been studied in patients who have previously failed therapy with a treatment regimen that includes OLYSIO or other HCV protease inhibitors.
Dosage and Administration
The recommended dose of OLYSIO is one capsule of 150 mg taken orally once daily with food. The type of food does not affect exposure to simeprevir. The capsule should be swallowed as a whole.
OLYSIO should be used in combination with peginterferon alfa and ribavirin. For peginterferon alfa and ribavirin specific dosage instructions, refer to their respective prescribing information.
Hepatic Impairment
No dose recommendation can be given for patients with moderate or severe hepatic impairment (Child-Pugh Class B or C) due to higher simeprevir exposures. In clinical trials, higher simeprevir exposures have been associated with increased frequency of adverse reactions, including rash and photosensitivity
The safety and efficacy of OLYSIO have not been studied in HCV-infected patients with moderate or severe hepatic impairment (Child-Pugh Class B or C). The combination of peginterferon alfa and ribavirin is contraindicated in patients with decompensated cirrhosis (moderate or severe hepatic impairment). The potential risks and benefits of OLYSIO should be carefully considered prior to use in patients with moderate or severe hepatic impairment.
WARNINGS AND PRECAUTIONS
Photosensitivity
Photosensitivity reactions have been observed with OLYSIO in combination with peginterferon alfa and ribavirin, including serious reactions which resulted in hospitalization. Photosensitivity reactions occurred most frequently in the first 4 weeks of treatment with OLYSIO in combination with peginterferon alfa and ribavirin, but can occur at any time during treatment. Photosensitivity may present as an exaggerated sunburn reaction, usually affecting areas exposed to light (typically the face, "V" area of the neck, extensor surfaces of the forearms, and dorsa of the hands). Manifestations may include burning, erythema, exudation, blistering, and edema.
Use sun protective measures and limit sun exposure during treatment with OLYSIO in combination with peginterferon alfa and ribavirin. Avoid use of tanning devices during treatment with OLYSIO in combination with peginterferon alfa and ribavirin. Discontinuation of OLYSIO should be considered if a photosensitivity reaction occurs and patients should be monitored until the reaction has resolved. If a decision is made to continue OLYSIO in the setting of a photosensitivity reaction, expert consultation is advised.
Rash
Rash has been observed in subjects receiving OLYSIO in combination with peginterferon alfa and ribavirin. Rash occurred most frequently in the first 4 weeks of treatment with OLYSIO in combination with peginterferon alfa and ribavirin, but can occur at any time during treatment. Severe rash and rash requiring discontinuation of OLYSIO have been reported. Most of the rash events in OLYSIO-treated patients were of mild or moderate severity. Patients with mild to moderate rashes should be followed for possible progression of rash, including the development of mucosal signs (e.g., oral lesions, conjunctivitis) or systemic symptoms. If the rash becomes severe, OLYSIO should be discontinued. Patients should be monitored until the rash has resolved.
Sulfa Allergy
OLYSIO contains a sulfonamide moiety. In subjects with a history of sulfa allergy (n=16), no increased incidence of rash or photosensitivity reactions has been observed. However, there are insufficient data to exclude an association between sulfa allergy and the frequency or severity of adverse reactions observed with the use of OLYSIO.
ADVERSE REACTIONS
The most common reported adverse reactions (greater than 20% of subjects) in subjects receiving the combination of OLYSIO with peginterferon and ribavirin and occurring with at least 3% higher frequency compared to subjects receiving placebo in combination with peginterferon alfa and ribavirin during the first 12 weeks of treatment were: rash (including photosensitivity), pruritus and nausea.
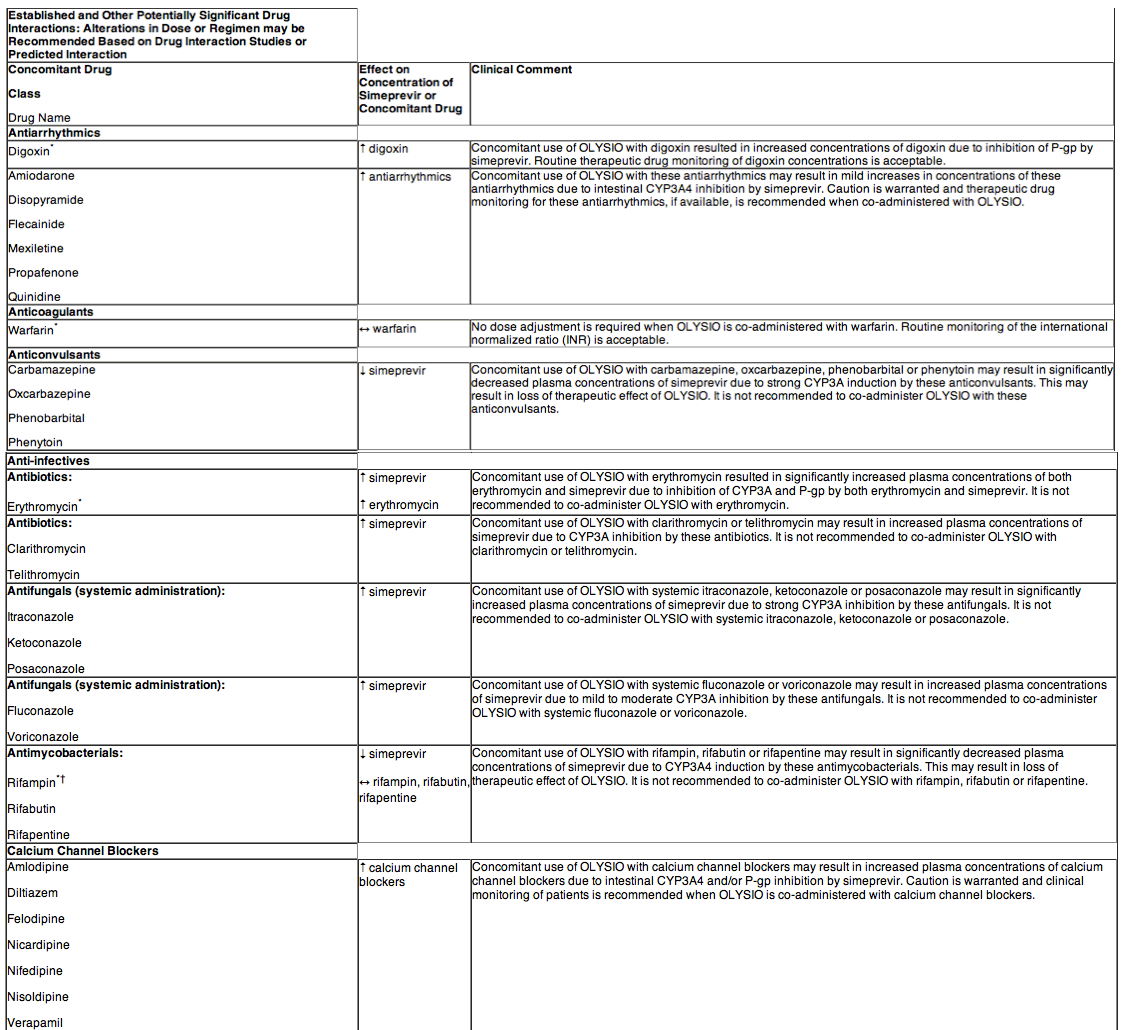
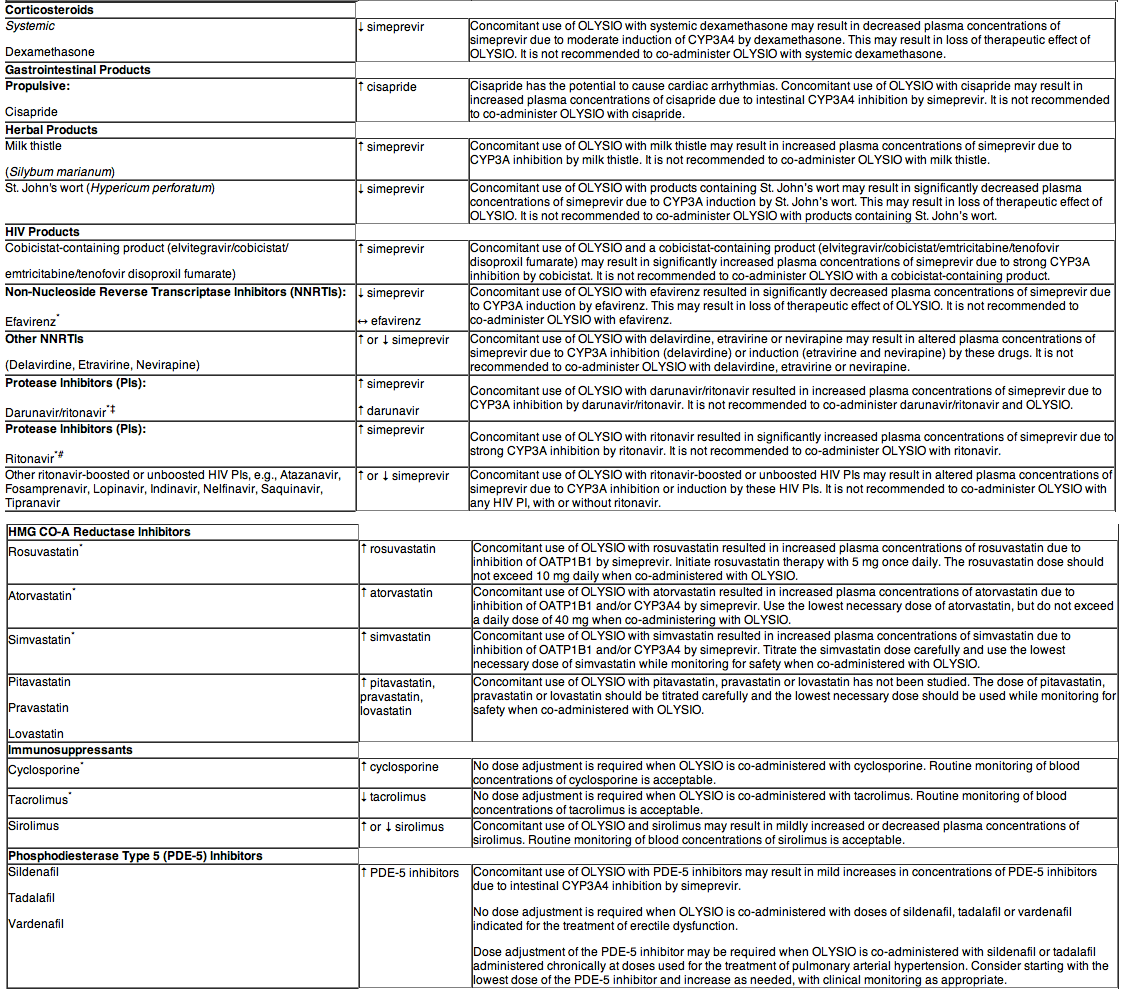
DRUG INTERACTIONS
Potential for OLYSIO to Affect Other Drugs
Simeprevir does not induce CYP1A2 or CYP3A4 in vitro. Simeprevir is not a clinically relevant inhibitor of cathepsin A enzyme activity.
Simeprevir mildly inhibits CYP1A2 activity and intestinal CYP3A4 activity, but does not affect hepatic CYP3A4 activity. Co-administration of OLYSIO with drugs that are primarily metabolized by CYP3A4 may result in increased plasma concentrations of such drugs. Simeprevir does not affect CYP2C9, CYP2C19 or CYP2D6 in vivo.
Simeprevir inhibits OATP1B1/3 and P-glycoprotein (P-gp) transporters. Co-administration of OLYSIO with drugs that are substrates for OATP1B1/3 and P-gp transport may result in increased plasma concentrations of such drugs.
Potential for Other Drugs to Affect OLYSIO
The primary enzyme involved in the biotransformation of simeprevir is CYP3A. Clinically relevant effects of other drugs on simeprevir pharmacokinetics via CYP3A may occur. Co-administration of OLYSIO with moderate or strong inhibitors of CYP3A may significantly increase the plasma exposure of simeprevir. Co-administration with moderate or strong inducers of CYP3A may significantly reduce the plasma exposure of simeprevir and lead to loss of efficacy. Therefore, co-administration of OLYSIO with substances that are moderate or strong inducers or inhibitors of CYP3A is not recommended.
CLINICAL STUDIES
The efficacy of OLYSIO in patients with HCV genotype 1 infection was evaluated in two Phase 3 trials in treatment-naïve subjects (trials QUEST 1 and QUEST 2), one Phase 3 trial in subjects who relapsed after prior interferon-based therapy (PROMISE) and one Phase 2b trial in subjects who failed prior therapy with peginterferon (Peg-IFN) and ribavirin (RBV) (including prior relapsers, partial and null responders) (ASPIRE). Prior relapsers were subjects who had undetectable HCV RNA at the end of prior IFN-based therapy and detectable HCV RNA during follow-up; prior partial responders were subjects with prior on-treatment greater than or equal to 2 log10 reduction in HCV RNA from baseline at Week 12 and detectable HCV RNA at the end of prior therapy with Peg-IFN and RBV; and null responders were subjects with prior on-treatment less than 2 log10 reduction in HCV RNA from baseline at Week 12 during prior therapy with Peg-IFN and RBV. Subjects in these trials had compensated liver disease (including cirrhosis), HCV RNA of at least 10000 IU/mL, and liver histopathology consistent with CHC.
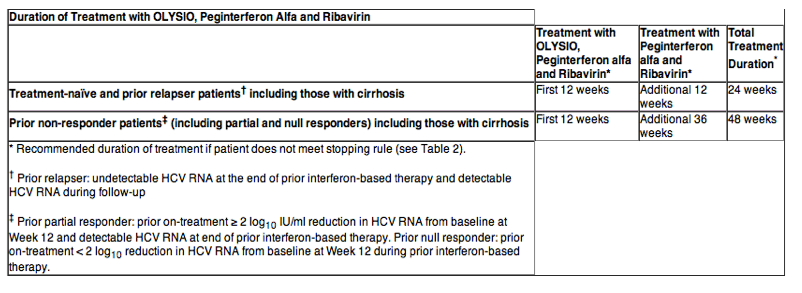
In subjects who were treatment-naïve and prior relapsers, the overall duration of treatment with Peg-IFN-alfa and RBV in the Phase 3 trials was response-guided. In these subjects, the planned total duration of HCV treatment was 24 weeks if the following on-treatment protocol-defined response-guided therapy (RGT) criteria were met: HCV RNA lower than 25 IU/mL (detectable or undetectable) at Week 4 AND undetectable HCV RNA at Week 12. Plasma HCV RNA levels were measured using the Roche COBAS® TaqMan® HCV test (version 2.0), for use with the High Pure System (25 IU/mL LLOQ and 15 IU/mL limit of detection). Treatment stopping rules for HCV therapy were used to ensure that subjects with inadequate on-treatment virologic response discontinued treatment in a timely manner.
SVR (virologic cure) was defined as undetectable HCV RNA 24 weeks after planned end of treatment (SVR24) in the Phase 2b trial and was defined as HCV RNA lower than 25 IU/mL detectable or undetectable 12 weeks after the planned end of treatment (SVR12) in the Phase 3 trials.
Treatment-Naïve Adult Subjects with HCV Genotype 1 Infection
The efficacy of OLYSIO in treatment-naïve patients with HCV genotype 1 infection was demonstrated in two randomized, double-blind, placebo-controlled, 2-arm, multicenter, Phase 3 trials (QUEST 1 and QUEST 2). The design of both trials was similar. All subjects received 12 weeks of once daily treatment with 150 mg OLYSIO or placebo, plus Peg-IFN-alfa-2a (QUEST 1 and QUEST 2) or Peg-IFN-alfa-2b (QUEST 2) and RBV, followed by 12 or 36 weeks of therapy with Peg-IFN-alfa and RBV in accordance with the on-treatment protocol-defined RGT criteria. Subjects in the control groups received 48 weeks of Peg-IFN-alfa-2a or -2b and RBV.
In the pooled analysis for QUEST 1 and QUEST 2, demographics and baseline characteristics were balanced between both trials and between the OLYSIO and placebo treatment groups. In the pooled analysis of trials (QUEST 1 and QUEST 2), the 785 enrolled subjects had a median age of 47 years (range: 18 to 73 years); 56% were male; 91% were White, 7% Black or African American, 1% Asian, and 17% Hispanic; 23% had a body mass index (BMI) greater than or equal to 30 kg/m2; 78% had HCV RNA levels greater than 800000 IU/mL; 74% had METAVIR fibrosis score F0, F1 or F2, 16% METAVIR fibrosis score F3, and 10% METAVIR fibrosis score F4 (cirrhosis); 48% had HCV genotype 1a, and 51% HCV genotype 1b; 29% had IL28B CC genotype, 56% IL28B CT genotype, and 15%IL28B TT genotype; 17% of the overall population and 34% of the subjects with genotype 1a virus had the NS3 Q80K polymorphism at baseline. In QUEST 1, all subjects received Peg-IFN-alfa-2a; in QUEST 2, 69% of the subjects received Peg-IFN-alfa-2a and 31% received Peg-IFN-alfa-2b.
Table 10 shows the response rates in treatment-naïve adult subjects with HCV genotype 1 infection. In the OLYSIO treatment group, SVR12 rates were lower in subjects with genotype 1a virus with the NS3 Q80K polymorphism at baseline compared to subjects infected with genotype 1a virus without the Q80K polymorphism.

In the pooled analysis of QUEST 1 and QUEST 2, 88% (459/521) of OLYSIO-treated subjects were eligible for a total treatment duration of 24 weeks. In these subjects, the SVR12 rate was 88% (405/459).
Seventy-eight percent (78%; 404/521) of OLYSIO-treated subjects had undetectable HCV RNA at Week 4 (RVR); in these subjects the SVR12 rate was 90% (362/404), while 8% (32/392) with undetectable HCV RNA at end of treatment had viral relapse.
SVR12 rates were higher for the OLYSIO treatment group compared to the placebo treatment group by sex, age, race, BMI, HCV genotype/subtype, baseline HCV RNA load (less than or equal to 800000 IU/mL, greater than 800000 IU/mL), METAVIR fibrosis score, and IL28B genotype. Table 11 shows the SVR rates by METAVIR fibrosis score.

SVR12 rates were higher for subjects receiving OLYSIO with Peg-IFN-alfa-2a or Peg-IFN-alfa-2b and RBV (88% and 78%, respectively) compared to subjects receiving placebo with Peg-IFN-alfa-2a or Peg-IFN-alfa-2b and RBV (62% and 42%, respectively) (QUEST 2).
Adult Subjects with HCV Genotype 1 Infection who Failed Prior Therapy
The PROMISE trial was a randomized, double-blind, placebo-controlled, 2-arm, multicenter, Phase 3 trial in subjects with HCV genotype 1 infection who relapsed after prior IFN-based therapy. All subjects received 12 weeks of once daily treatment with 150 mg OLYSIO or placebo, plus Peg-IFN-alfa-2a and RBV, followed by 12 or 36 weeks of therapy with Peg-IFN-alfa-2a and RBV in accordance with the protocol-defined RGT criteria. Subjects in the control group received 48 weeks of Peg-IFN-alfa-2a and RBV.
Demographics and baseline characteristics were balanced between the OLYSIO and placebo treatment groups. The 393 subjects enrolled in the PROMISE trial had a median age of 52 years (range: 20 to 71 years); 66% were male; 94% were White, 3% Black or African American, 2% Asian, and 7% Hispanic; 26% had a BMI greater than or equal to 30 kg/m2; 84% had HCV RNA levels greater than 800000 IU/mL; 69% had METAVIR fibrosis score F0, F1 or F2, 15% METAVIR fibrosis score F3, and 15% METAVIR fibrosis score F4 (cirrhosis); 42% had HCV genotype 1a, and 58% HCV genotype 1b; 24% had IL28B CC genotype, 64% IL28B CT genotype, and 12% IL28B TT genotype; 13% of the overall population and 31% of the subjects with genotype 1a virus had the NS3 Q80K polymorphism at baseline. The prior IFN-based HCV therapy was Peg-IFN-alfa-2a /RBV (68%) or Peg-IFN-alfa-2b/RBV (27%).
Table 12 shows the response rates for the OLYSIO and placebo treatment groups in adult subjects with HCV genotype 1 infection who relapsed after prior interferon-based therapy. In the OLYSIO treatment group, SVR12 rates were lower in subjects infected with genotype 1a virus with the NS3 Q80K polymorphism at baseline compared to subjects infected with genotype 1a virus without the Q80K polymorphism.

In PROMISE, 93% (241/260) of OLYSIO-treated subjects were eligible for a total treatment duration of 24 weeks. In these subjects, the SVR12 rate was 83% (200/241).
Seventy-seven percent (77%; 200/260) of OLYSIO-treated subjects had undetectable HCV RNA at Week 4 (RVR); in these subjects the SVR12 rate was 87% (173/200), while 13% (25/196) with undetectable HCV RNA at end of treatment had viral relapse.
SVR12 rates were higher for the OLYSIO treatment group compared to the placebo treatment group by sex, age, race, BMI, HCV genotype/subtype, baseline HCV RNA load (less than or equal to 800000 IU/mL, greater than 800000 IU/mL), prior HCV therapy, METAVIR fibrosis score, and IL28B genotype. Table 13 shows the SVR rates by METAVIR fibrosis score.

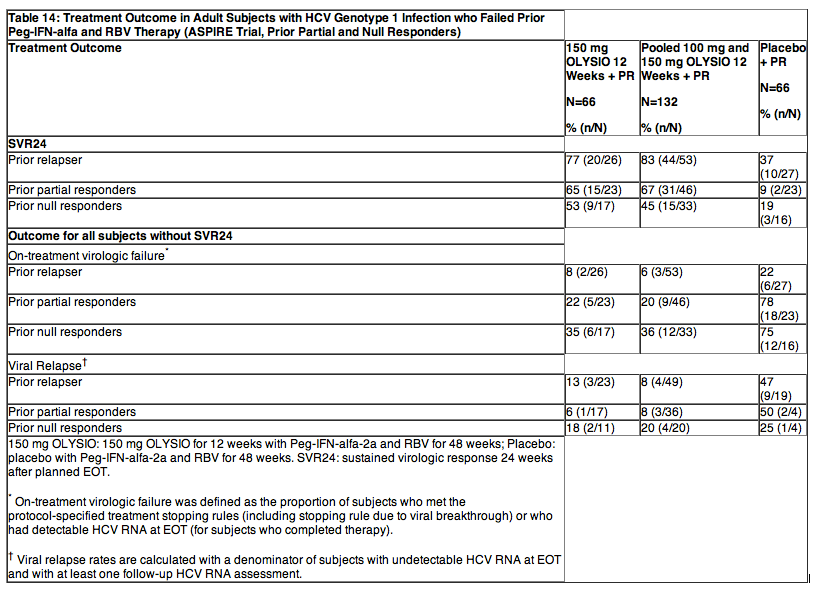
The ASPIRE trial was a randomized, double-blind, placebo-controlled, 7-arm, Phase 2b trial in subjects with HCV genotype 1 infection, who failed prior therapy with Peg-IFN-alfa and RBV (including prior relapsers, partial responders or null responders). Subjects received 12, 24 or 48 weeks of 100 mg or 150 mg OLYSIO in combination with 48 weeks of Peg-IFN-alfa-2a and RBV, or 48 weeks of placebo in combination with 48 weeks of Peg-IFN-alfa-2a and RBV.
Demographics and baseline characteristics were balanced between the OLYSIO and placebo treatment groups. The 462 subjects enrolled in the ASPIRE trial had a median age of 50 years (range: 20 to 69 years); 67% were male; 93% were White, 5% Black or African American, and 2% Asian; 25% had a BMI greater than or equal to 30 kg/m2; 86% had HCV RNA levels greater than 800000 IU/mL; 63% had METAVIR fibrosis score F0, F1, or F2, 19% METAVIR fibrosis score F3, and 18% METAVIR fibrosis score F4 (cirrhosis); 41% had HCV genotype 1a, and 58% HCV genotype 1b; 18% hadIL28B CC genotype, 65% IL28B CT genotype, and 18% IL28B TT genotype (information available for 328 subjects); 12% of the overall population and 27% of the subjects with genotype 1a virus had the NS3 Q80K polymorphism at baseline. Forty percent (40%) of subjects were prior relapsers, 35% prior partial responders, and 25% prior null responders following prior therapy with Peg-INF-alfa and RBV. One hundred ninety-nine subjects received OLYSIO 150 mg once daily (pooled analysis) of which 66 subjects received OLYSIO for 12 weeks and 66 subjects received placebo in combination with Peg-IFN-alfa and RBV.
Table 14 shows the response rates for the OLYSIO and placebo treatment groups in prior relapsers, prior partial responders and prior null responders.

In prior partial responders, SVR24 rates in subjects receiving OLYSIO with Peg-IFN-alfa and RBV were 47% and 77% in subjects with HCV genotype 1a and 1b, respectively, compared to 13% and 7%, respectively, in subjects receiving placebo with Peg-IFN-alfa and RBV. In prior null responders, SVR24 rates in subjects receiving OLYSIO with Peg-IFN-alfa and RBV were 41% and 47% in subjects with HCV genotype 1a and 1b, respectively, compared to 0% and 33%, respectively, in subjects receiving placebo with Peg-IFN-alfa and RBV.
SVR24 rates were higher in the OLYSIO-treated subjects compared to subjects receiving placebo in combination with Peg-IFN-alfa and RBV, regardless of HCV geno/subtype, METAVIR fibrosis score, and IL28B genotype.
The complete label will be available soon at Drugs@FDA
Richard Klein
Office of Special Health Issues
Food and Drug Administration
Kimberly Struble
Division of Antiviral Drug Products
Food and Drug Administration
|
|
| |
| |
|
|
|