| |
Randomized Placebo-Controlled Study of the Safety, Tolerability, Antiviral Activity, and Pharmacokinetics of 10-Day Monotherapy With BMS-986001, a Novel HIV NRTI, in Treatment-Experienced HIV-1-Infected Subjects & BMS626529 Attachment Inhibitor
|
| |
| |
Download the PDF here
ResisWksp/2013: Frequency of amino acid changes associated with resistance to attachment inhibitor BMS-626529 in R5- and X4-tropic HIV-1 subtype B (06/12/13)
IAS/2012: THE HIV NRTI BMS-986001 DOES NOT DEGRADE MITOCHONDRIAL DNA IN LONG TERM PRIMARY CULTURES OF CELLS ISOLATED FROM HUMAN KIDNEY, MUSCLE AND SUBCUTANEOUS FAT
http://www.natap.org/2012/IAS/IAS_21.htm
Absence of renal and bone toxicity in non-clinical studies of BMS-986001, a nucleoside reverse transcriptase inhibitor (NRTI) of human immunodeficiency virus (HIV).......http://www.natap.org/2012/IAS/IAS_17.htm
Good Virologic Response in 10-Day Study of Festinavir, a New NRTI.....http://www.natap.org/2012/pharm/Pharm_02.htm
Antiviral activity, exposure-response, and resistance analyses of monotherapy with the novel HIV nucleoside reverse transcriptase inhibitor (NRTI) BMS-986001 in antiretroviral treatment-experienced subjects.......http://www.natap.org/2012/pharm/Pharm_11.htm
Lack of Resistance Development to the HIV-1 Attachment Inhibitor BMS-626529 During Short-Term Monotherapy with its Prodrug BMS-663068
http://www.natap.org/2012/CROI/croi_39.htm
The in vitro cross-resistance profile of the NRTI BMS-986001 against known NRTI resistance mutations
http://www.natap.org/2012/ResisWksp/ResisWksp_07.htm
Pharmacodynamics, Safety, and Pharmacokinetics of BMS-663068, an Oral HIV-1 Attachment Inhibitor in HIV-1-Infected Subjects.......http://www.natap.org/2012/HIV/090712_02.htm
Activity of the HIV-1 attachment inhibitor BMS-626529 against HIV-1 envelopes resistant to other entry inhibitors
http://www.natap.org/2012/IAS/IAS_20.htm
Frequency of amino acid changes associated with resistance to attachment inhibitor BMS-626529 in R5- and X4-tropic HIV-1 subtype B
http://www.natap.org/2013/ResisWksp/ResisWksp_10.htm
JAIDS Journal of Acquired Immune Deficiency Syndromes: July 1 2013
Cotte, Laurent MD*; Dellamonica, Pierre MD; Raffi, Francois MD, PhD; Yazdanpanah, Yazdan MD, PhD; Molina, Jean-Michel MD; Boue, Francois MD; Urata, Yasuo MSc#; Chan, H. Phyllis PhD**; Zhu, Li PhD**; Chang, Ih PhD**; Bertz, Richard PhD**; Hanna, George J. MD**; Grasela, Dennis M. PharmD, PhD**; Hwang, Carey MD, PhD**
Presented in part at the 50th Annual Interscience Conference on Antimicrobial Agents and Chemotherapy, Boston, MA, September 12-15, 2010 (Abstract H-933) and at the 13th International Workshop on Clinical Pharmacology of HIV Therapy, Barcelona, Spain, April 16-18, 2012 (poster O_06).
Abstract
Objective: To investigate the safety, tolerability, pharmacokinetics, and antiviral activity of BMS-986001 (a nucleoside reverse transcriptase inhibitor) in treatment-experienced, HIV-1-infected subjects not exposed to antiretroviral treatment in the previous 3 months.
Methods: Thirty-two HIV-1-infected subjects were randomized (3:1) to receive BMS-986001 or placebo once daily for 10 days in this double-blind, placebo-controlled, dose-escalating monotherapy phase IIa study. There were 4 treatment groups (100, 200, 300, and 600 mg, all once daily) of 8 subjects each (BMS-986001, n = 6/placebo n = 2).
Results: BMS-986001 was generally well tolerated, with no discontinuations due to adverse events and no deaths occurring. Adverse events were experienced by 22 of 24 BMS-986001-treated subjects and did not seem to be dose related. The majority were mild and considered unrelated or unlikely to be related to the study drug. The pharmacokinetics of BMS-986001 were dose proportional. Median decrease in plasma HIV-1 RNA from baseline to day 11 was 0.97, 1.15, 1.28, and 1.15 log10 copies/mL for BMS-986001 at 100, 200, 300, and 600 mg, respectively. Plasma area under the curve correlated with the antiviral activity of BMS-986001, indicating that area under the curves produced by 100-600 mg doses were on the upper end of the exposure-response curve. One subject with a single thymidine analog mutation at baseline responded well to BMS-986001.
Conclusions: Administration of BMS-986001 for 10 days resulted in substantial decreases in plasma HIV-1 RNA levels for all dose groups and was generally well tolerated. These data support continued clinical development of BMS-986001 at a dose of 100 mg, once daily or greater.
INTRODUCTION
Nucleoside/nucleotide reverse transcriptase inhibitors (NRTIs) remain a cornerstone of combination antiretroviral therapy (cART) for HIV. Current treatment guidelines recommend that cART regimens for treatment-naive patients consist of 2 NRTIs plus 1 other active agent, a non-NRTI, a protease inhibitor or an integrase inhibitor.1 Despite the number of NRTIs available, and their wide use, there are ongoing concerns regarding the established and potential long-term toxicities of the class. Well-established toxicities include anemia [zidovudine (AZT)], pancreatitis [didanosine (ddI)], and other issues related to mitochondrial toxicity, including peripheral neuropathy [ddI and stavudine (d4T)], lipoatrophy (d4T and AZT), and metabolic acidosis (mostly with d4T, ddI, and AZT).1 As a result of these toxicities, many NRTIs are now only used under special circumstances.1 Lamivudine (3TC) and emtricitabine (FTC) have a favorable safety profile but cannot be used together because of a shared mechanism of action and a similar resistance profile. Major HIV-1 treatment guidelines currently recommend combinations of tenofovir disoproxil fumarate (TDF) or abacavir (ABC) with FTC or 3TC as part of initial HIV-1 treatment regimens.1-3 ABC is not always preferred because it is associated with hypersensitivity reaction, and there is conflicting evidence regarding its effect on cardiovascular risk.1,3-6 Although TDF is preferred because of favorable tolerability and efficacy profile, concerns regarding nephrotoxicity decreased bone mineral density and increased risk of fractures remain.1,7,8
The life expectancy for HIV-infected individuals has increased notably in the last decade,9 resulting in a population of increasing age and disease chronicity. This, together with the earlier start of cART treatment recommended by clinical guidelines,1 means that many patients receive antiretroviral therapies for increasingly long durations.
The aforementioned concerns highlight a need for novel NRTIs with potent anti-HIV-1 activity, a good tolerability profile (similar or better than currently available antiretroviral agents), long-term safety (in terms of bone mineral density loss/decline in renal function), and limited cross-resistance to existing NRTIs. Furthermore, a convenient regimen to improve adherence (eg, once daily dosing or fixed dose combinations) is an increasingly important factor in the development of new NRTIs. Thus, pharmacokinetic (PK) profile and drug metabolism are important considerations.
BMS-986001 is a thymidine NRTI that has been developed to maintain the in vitro antiviral activity demonstrated by other NRTIs without the associated toxicity concerns. BMS-986001 is a novel analog of d4T, which, in preliminary in vitro experiments, demonstrated potent antiviral activity and reduced mitochondrial toxicity compared with d4T.10 It displayed reduced inhibition of host DNA polymerases compared with other thymidine analogs,11 and there was evidence of a higher genetic barrier to resistance than its progenitor d4T.12 Furthermore, BMS-986001 has demonstrated activity against a number of HIV-1 subtypes (Bristol-Myers Squibb, unpublished data, 2011) and against HIV-1 isolates with certain NRTI-associated mutations, such as K65R, L74V, and Q151M mutations.13 Viruses with thymidine analog mutations (TAMs) conferring reduced susceptibility to BMS-986001 were generally cross-resistant to many other NRTIs (Bristol-Myers Squibb, unpublished data, 2011). M184V induced low-level fold change (2- to 3-fold) in susceptibility to BMS-986001.12,13
The safety, tolerability, PK, and pharmacodynamic effects of BMS-986001 were evaluated in a 10-day monotherapy trial in treatment-experienced HIV-1-infected subjects who were not exposed to any antiretroviral treatment in the previous 3 months.
RESULTS
Subject Disposition
A total of 32 subjects (median time since diagnosis, 12.0 years) were randomized and received study drug. All subjects were previously treated with NRTIs (see Table S1, Supplemental Digital Content, http://links.lww.com/QAI/A424, which shows previous instances of NRTI treatment at baseline). Baseline demographics and disease characteristics were generally well balanced between groups (Table 1). At baseline, median plasma HIV-1 viral RNA ranged from 4.0 to 4.8 log10 copies/mL and median CD4+ T-cell count from 306.5 to 572.0 cells/μL. No subject received prohibited concomitant therapy. All 32 subjects were included in the safety and antiviral activity assessment groups. Two subjects were excluded from the PK data sets: one who did not receive study drug on day 10 was excluded from the day 10 summary and one who received the incorrect dose on day 1 was excluded from the day 1 summary.
Safety and Tolerability
A summary of AEs is presented in Table 2. A total of 27 (84%) subjects reported 95 treatment-emergent AEs [82 (86.3%) grade 1; 11 (11.6%) grade 2; 2 (2.1%) grade 3], of which 30 were considered possibly study drug related, 7 unlikely to be related to study drug, and 58 unrelated to study drug (as assessed by the investigator). A greater proportion of BMS-986001-treated subjects reported grade 2 and 3 AEs [6/24 (25%)] compared with placebo [1/8 (12%)]; subjects may have experienced more than 1 AE. There were no grade 4 events. The median time between first dose and AE occurrence was 9 (0-23) days and 7 (0-23) days for subjects receiving BMS-986001 and placebo, respectively. AEs reported by subjects receiving BMS-986001 did not seem to be dose related; however, small subject numbers must be taken into consideration. The most common AEs (>10% of subjects) in the BMS-986001 treatment groups were abdominal pain (n = 5), lymphadenopathy (n = 4), nausea (n = 3), headache (n = 3), and fatigue (n = 3). In the placebo group, the most common AEs were nausea (n = 2) and increased blood lactic acid (n = 2).
The majority of AEs resolved before study discharge. One AE of grade 3 anxiety (100-mg group) and 1 AE of grade 1 fatigue (300-mg group) were unresolved at study discharge and were considered to be related to the study drug. The subject with grade 3 anxiety had a history of anxiety and also experienced grade 3 paranoia.
Two subjects in the 600-mg group each reported 1 serious AE, neither considered study drug related; streptococcal septicemia and cytomegalovirus primary infection. Three subjects experienced a grade 3/4 laboratory abnormality, none of which required intervention; decreased platelets (100-mg group), elevated serum potassium (200-mg group), and elevated LDL (placebo). The abnormality of decreased platelets occurred on day 17 (4.5-fold decrease from day 11) and returned to normal on day 24. There were no changes in other hematologic values for this subject; the decrease in platelets was probably because of platelet clumping. Elevated serum potassium (6.9 mEq/L) occurred on day 1 before dosing and returned to normal (4.3 mEq/L) on day 3. The elevation in LDL cholesterol was present at screening and remained elevated throughout. There were no clinically important ECG, vital sign, and other lipid or physical examination changes during follow-up.
Pharmacokinetics
Individual PK parameters for BMS-986001 are summarized by treatment in Table 3. At all doses, BMS-986001 was rapidly absorbed; maximum plasma BMS-986001 concentration was observed approximately 1-2 hours postdose in all groups. PK parameters were similar on days 1 and 10. There was no significant accumulation at any of the doses studied. The increase in exposure for Cmax and AUCtau to BMS-986001 over the dose range of 100-600 mg once daily for 10 days was dose proportional based on the power model: slope estimate (90% CI) for AUCtau at day 10 = 1.025 (0.913 to 1.137) and Cmax at day 10 = 0.986 (0.844 to 1.129).
Antiviral Activity
At day 11, the median decrease in plasma HIV-1 RNA levels was 1.20 log10, for all BMS-986001 doses combined (Fig. 1A). By day 11, the median (range) decrease was 0.97 (1.3 to -0.2), 1.15 (1.3 to 0.4), 1.28 (1.6 to 1.2), 1.15 (1.9 to -0.1), and 0.11 (0.3 to -0.2) log10 copies/mL for BMS-986001 treatment groups 100, 200, 300, and 600 mg and placebo, respectively (see Figure S1, Supplemental Digital Content, http://links.lww.com/QAI/A424, for median change in plasma HIV-1 RNA level over time according to treatment group). Individual response variability was observed (Fig. 1B). Three subjects had decreased or no response (defined as <0.5 log10 copies/mL decline in plasma HIV-1 RNA by day 11) to BMS-986001. These 3 subjects had no NRTI-associated resistance mutations (including TAMs) at baseline. Previous treatments for each of the 3 patients were as follows-subject 1: AZT, 3TC, lopinavir/ritonavir, TDF/FTC, and atazanavir/ritonavir; subject 2: AZT, ddC, indinavir, and nevirapine; and subject 3: AZT and 3TC. Two of these 3 subjects had predose HIV-1 RNA <5000 copies/mL (3650 copies/mL for subject 1 and 307 copies/mL for subject 3).
Increases from baseline in median CD4+ T-cell counts were recorded in all BMS-986001 groups at day 11 (71.00-178.50 cells/μL) and all BMS-986001 groups except for the 600-mg group at day 24 (-20.00 to 145.50 cells/μL) (Fig. 2A). There was considerable variability in individual CD4+ T-cell response (Fig. 2B).
Exposure-Response Analysis
The exposure--response relationship between BMS-986001 steady-state exposure (AUCtau) and change in HIV-1 RNA levels on day 11 was explored. An Emax model provided adequate fit to the data (see Figure S2, Supplemental Digital Content, http://links.lww.com/QAI/A424, which demonstrates the relationship between BMS-986001 AUCtau on day 10 and plasma HIV-1 RNA change on day 11 from day 1, after 10 days of monotherapy with BMS-986001 from 100 to 600 mg). The relationship between BMS-986001 plasma exposure (AUCtau) and change in log10 plasma HIV-1 RNA levels on day 11 from day 1 was explored using nonlinear regression methods. An Emax model was applied to the data:
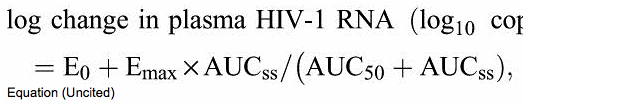
where E0 = effect in absence of BMS-986001, Emax = maximum effect, AUCss = AUC at steady state, and AUC50 = AUC required to achieve 50% of maximum reduction in HIV-1 RNA level.
This result suggests that the BMS-986001 plasma AUC is correlated with its antiviral activity in humans. The AUC50 estimated from the analysis was 3485 ng·h/mL, and indicates that AUC values produced by doses from 100 to 600 mg were on the upper end of the exposure-response curve. Two nonresponders (<0.5 log10 decline in copy number; 1 subject from the 100-mg group and 1 subject from the 600-mg group) were excluded from the analysis as although their viral load met entry criteria (>5000 copies/mL) at screening (1-21 days before initialization of treatment), by the time of randomization into the study, their viral load was <5000 copies/mL and hence did not meet entry criteria. Exclusion of these 2 nonresponders improved model fitting and precision of parameter estimates.
Resistance Analysis
At baseline, only 1 subject had an NRTI resistance mutation detectable by population sequencing [see Table S2, Supplemental Digital Content, http://links.lww.com/QAI/A424, which shows all amino acid changes from wild-type in protease and reverse transcriptase at days 1 (baseline), 11, and 17]. This subject was in the 600-mg group and had T215T/S on day 1, which was not present on days 11 or 17. Two subjects in the 300-mg group had K70K/R and K219K/R, respectively, on day 17, which were not present on days 1 or 11. All 3 subjects responded well to BMS-986001. For subjects with the T215T/S, K70K/R, or K219K/R mutations, decline in HIV-1 RNA from baseline to day 11 was 1.91, 1.42, or 1.58 log10 copies/mL, respectively. Three subjects had a decreased response to BMS-986001 (<0.5 log10 decrease in HIV-1 RNA) on day 11; analysis showed that there were no NRTI-associated resistance mutations at baseline. Despite a history of previous treatment and failure to 3TC and/or FTC in several subjects, the M184V mutation was not detected at any time in any subject.
DISCUSSION
These results represent the first report of the safety and antiviral activity of the novel HIV-1 NRTI BMS-986001 in HIV-1-infected subjects. Overall, BMS-986001 was generally well tolerated in this study. AEs did not seem to have a dose-dependent relationship and the majority were mild and deemed not related or unlikely related to study treatment. No AEs necessitated study drug discontinuation. BMS-986001 was rapidly absorbed at all doses studied, reaching Cmax in approximately 1-2 hours. Mean plasma concentration-time profiles for BMS-986001 on days 1 and 10 were largely overlapping for all dose groups, indicating no significant accumulation in BMS-986001 systemic exposures following once daily dosing. BMS-986001 demonstrated a linear dose-exposure relationship across the dose ranges studied following 10 days of once daily dosing. The increases in plasma Cmax and AUC were generally proportional to increases in dose. For all doses studied, BMS-986001 was rapidly absorbed and results were consistent with single-ascending-dose study results in uninfected subjects (Bristol-Myers Squibb, unpublished data, 2011).
BMS-986001 is metabolized intracellularly to its active triphosphate form. In vitro studies in CEM cells (CD4+ T-cell line) showed that BMS-986001 triphosphate had an intracellular T1/2 of approximately 9.7 hours.17 The intracellular concentration of the active component over time in HIV-1-infected subjects and correlation with the extent of reduction in HIV-1 RNA is currently being evaluated.
Substantial decreases in plasma HIV-1 RNA were recorded for the majority of subjects, and the decrease at day 11 with BMS-986001 compared favorably with observations with other potent NRTIs, including TDF.18 It should be noted, however, that there are differences in the study populations between this TDF monotherapy study18 and the current study, including for example, the degree of previous exposure to antiretrovirals. A similar median decrease in plasma HIV-1 RNA of ≥1 log10 was observed in all arms; therefore, antiviral activity at doses of 100-600 mg is roughly comparable. Increases from baseline in median CD4+ T-cell counts were also recorded in all BMS-986001 groups (except the 600-mg group) at days 11 and 24. However, high variability among individual subjects was observed within each dose group, including a fall in median CD4+ T-cell counts in 2 subjects. A possible contributor to the variation in CD4+ T-cell responses detected was the inadequate conservation of cells during transport and storage. The circadian rhythm of leukocytes and variation in complete blood cell count according to minor infections may also have contributed.
Exposure-response analysis suggested a correlation between BMS-986001 plasma AUCtau and antiviral activity, with doses from 100 to 600 mg resulting in geometric mean values for plasma exposure in excess of the EC50 value for decline in plasma HIV-1 RNA.
Three subjects had a decreased response to BMS-986001 (<0.5 log10 decrease in HIV-1 RNA) on day 11, none of whom had NRTI-associated resistance mutations at baseline or on day 11. The subjects had comparable PK to other subjects in their respective dose groups. In 1 subject, HIV-1 RNA was 15,000 copies/mL at screening, but 307 copies/mL at baseline, which contributed to the low change from baseline estimated for BMS-986001. Three other subjects who had TAMs responded well to BMS-986001. These data suggest that the decreased response in some subjects is unlikely to be due to known NRTI resistance-associated mutations. A limitation of the study methodology, however, is that the assay used for resistance analysis did not cover the reverse transcriptase connection or RNase H domains of the HIV-1 genome. Historical genotype data were also not available. In addition, some doses were administered at home; thus, subject adherence and its impact on individual virologic response in this study cannot be determined. Variability in technical procedures may also have contributed.
In conclusion, BMS-986001 is a once daily NRTI with potent antiviral activity and linear PK. It was generally well tolerated when administered for 10 days in treatment-experienced HIV-1-infected subjects. These data support the further clinical development of BMS-986001 in cART; a phase 2b study in treatment-naive HIV-1-infected patients (NCT01489046) is ongoing. Doses of 100, 200, and 400 mg once daily are being evaluated as they are projected to produce a wide range of exposures that will allow for differentiation of safety and efficacy and provide critical data for the optimal selection of a phase 3 dose.
METHODS
Study Design
A randomized, double-blind, placebo-controlled, dose-escalating, 10-day monotherapy study was conducted in treatment-experienced HIV-1-infected subjects at 6 centers in France between December 2008 and September 2009. Four treatment groups (100, 200, 300, and 600 mg, all once daily) of 8 subjects each were enrolled. Within each group, subjects were randomized by a computer-generated randomization scheme (3:1 ratio) to receive BMS-986001 or placebo once daily for 10 days (BMS-986001, n = 6/placebo, n = 2). An independent Data Safety Monitoring Board was responsible for authorization of dose escalation and met 3 times during the study, once after each of the 100-, 200-, and 300-mg groups had completed treatment. A screened subject was included once and received 1 dose level. Study drug was administered as a single dose in the morning under fasting conditions. No food was allowed until 1 hour postdose. Doses were administered in the clinic on days 1, 2, 3, 5, and 10 and at home on the remaining days. Treatment allocation was performed by CRID Pharma according to a randomization list provided by the Contract Research Organization that remained blinded until database lock. There were 2 protocol amendments, one to test total cholesterol, low-density lipoprotein (LDL), gamma-glutamyl transpeptidase (γGTP), and triglycerides in the fasting state, and the other for amylase testing. The study protocol, its amendments, and informed consent documents were approved by the French Regulatory Authority (AFSSAPS) and by the independent ethics committee of the coordinating center in Lyon, France. Written informed consent was obtained from all participants. The studies were performed in accordance with International Conference on Harmonization Good Clinical Practice guidelines.
Study Population
Subjects underwent screening evaluations up to 21 days before administration of study medication. Eligible subjects were HIV-1-infected adults, male or female (aged 18-60 years), with plasma HIV-1 RNA ≥5000 copies/mL (using standard approved HIV-1 RNA assays) and CD4+ T-cell counts ≥250 cells/mL stable within 3-6 months. Subjects were antiretroviral experienced but with no antiretroviral therapy within 3 months before screening and no indication for antiretroviral therapy according to guidelines at the time of the study.14 Concomitant therapy with nucleoside analogs and antiretroviral agents other than BMS-986001 was prohibited. Subjects did not receive concomitant therapy (prescription, over-the-counter or herbal) for the treatment of specific clinical events, unless prescribed by the investigator. Exclusion criteria were hepatitis B or C co-infection, history of hepatic or abnormal liver function tests (defined as ≥2.5 x upper limit of normal), renal disease, or calculated creatinine clearance ≤80 mL/min (calculated using Cockroft-Gault15) at screening or history of significant hematologic disease.
Evaluations
Safety and Tolerability
Adverse events (AEs) were recorded throughout. Clinical laboratory evaluations (hematology, blood chemistry, and urinalysis) were performed at screening and on days 1, 3, 5, 11, 17, and 24. Echocardiograms (ECGs) were performed at screening and on days 1 (predose and 2 hours postdose) and 10 (2 hours postdose). Physical examinations and measurement of vital signs were performed at screening and on days 1, 5, 10, 12 (physical examination only), 17, and 24.
Antiviral Activity
Plasma HIV-1 RNA level was determined at screening and on days 1 (predose), 3, 5, 11, 17, and 24. CD4+ T-cell count was determined at screening and on days 1 (predose), 11, and 24.
Pharmacokinetics
Blood samples for PK analysis were collected predose and at 1, 2, 4, 9, 12, and 24 hours postdose on days 1 and 10. Samples were also collected predose on days 3 and 5 and on day 11 for analysis of trough concentration and on the morning of day 12. BMS-986001 concentration was assayed by validated liquid chromatography dual mass spectrometry. The lower limit of quantification for BMS-986001 was 1.00 ng/mL with overall coefficient of variance ≤8.1%. PK variables assessed included maximum observed plasma concentration (Cmax), time to Cmax (Tmax), area under the concentration-time curve in 1 dosing interval (AUCtau), terminal plasma half-life after the last dose administered (T1/2), and apparent total body clearance (CLT/F) in the fasted state. The PK parameters of BMS-986001 were derived from the plasma concentration-time profiles via noncompartmental methods using WinNonlin (version 5.0; Mountain View, CA). Actual sampling times were used for the analysis.
Resistance Analysis
HIV-1 drug resistance genotype was assessed on days 1 (predose), 11, and 17 using standard, commercially available testing procedures. This assay did not cover the reverse transcriptase connection or RNase H domains. Genotype data on subjects before enrollment in the study are not available. Resistance analysis was performed at Monogram Biosciences (San Francisco, CA).
Statistical Analysis
The primary objective was to evaluate the safety and tolerability of BMS-986001. The secondary objectives were to assess the PK profile of multiple doses of BMS-986001, evaluate the antiviral activity of BMS-986001 compared with placebo (change from baseline in plasma HIV-1 RNA levels), and evaluate the resistance profile of BMS-986001.
A sample size of 6 subjects in each dose group resulted in ≥80% probability of observing ≥1 occurrence of an event with underlying rate of ≥24% at a given dose level and ≥80% probability of observing a median HIV-1 RNA decrease of ≥0.8 log10 at a given dose level if the true rate of achieving such decrease is at least 60%. The statistical analysis was performed using the SAS software for Windows, version 9.2 (SAS Institute Inc., Cary, NC). Safety analysis dataset: all subjects who received ≥1 dose of study drug; PK analysis dataset: all subjects who received ≥1 of study drug and provided evaluable plasma concentration versus time profiles.
AEs were classified as pretreatment (onset date/time before date/time of the first study medication) or treatment-emergent (onset date/time on or after date/time of the first study medication). Frequency distribution of subjects with AEs was provided by treatment and coded according to MedDRA version 12.0, using preferred term and corresponding system organ class. The maximal severity grade and relationship to study drug was assessed by the investigator. Summary statistics were provided for vital signs, ECGs, and safety laboratory assessment data.
A descriptive analysis of plasma concentration at each time point and PK parameters of BMS-986001 on days 1 and 10 was performed. Individually, for days 1 and 10, statistical testing of dose proportionality of BMS-986001 in plasma for AUCtau and Cmax was performed using the power model of Gough et al.16 A mixed model, using PROC MIXED in SAS, was computed with the natural log-transformed PK parameter as a dependent variable and the natural log-transformed dose as a fixed effect. Within this model, the 90% confidence interval (CI) on the slope was calculated. Dose proportionality was concluded if the 90% CI for the slope estimate was completely contained within the range 0.8 to 1.25.
Summary statistics by treatment and time point were provided for CD4+ T-cell counts and log10-transformed plasma HIV-1 RNA levels. Baseline was defined as measurements obtained predose on day 1. Exploratory analysis of changes from baseline was conducted to compare each dose with placebo using Mann-Whitney U test. Exploratory analysis of Spearman rank correlation coefficients between dose level and difference from baseline in plasma HIV-1 RNA and CD4+ T-cell counts at day 11 was performed.
An exposure-response analysis was performed to explore the relationship between AUCtau on day 10 for BMS-986001 and day 11 change from baseline in log10 plasma HIV-1 RNA levels. Nonlinear regression modeling was performed using WinNonlin. Data on HIV-drug resistance genotype were listed for subjects with any change from baseline.
| |
| |
| |
|
|
|