| |
Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2: new research insight into HCV entry in developing HCV vaccine and inhibitors
|
| |
| |
Download the PDF here
Nature Feb 10 2014
"These results provide unprecedented insights into HCV entry and will assist in developing an HCV vaccine and new inhibitors."
http://blogs.dickinson.edu
Feb 23 2014
Scientists from Rutgers University and other institutions recently characterized a protein on the surface of Hepatitis C virus (HCV) and published their research in the journal Nature on February 19. Discovering this new information about may help to lead to the development of a vaccine for this debilitating virus, which affects an astounding 160 million people worldwide.
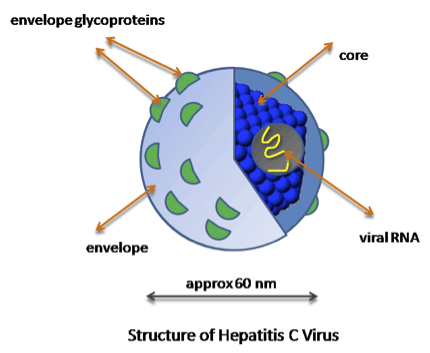
The structure of Hepatitis C virus. The E2 protein is one of the envelope glycoproteins referred to in the figure, which helps the virus fuse to cells and mediates entry.
The protein, called E2, is what scientists call a viral surface protein, or a protein that is present on the outer shell of viruses. In most cases, these proteins serve to help viruses interact with receptors on human cells in order to gain entry into the cells and infect them. Learning the extensive biochemistry of these types of proteins can help scientists to better understand how viruses infect cells, and in turn can be useful for producing vaccines and other anti-viral drugs. Vaccines often utilize viral surface proteins in order to prime the immune system, so that when a person is actually infected with a virus like HCV the body already has antibodies (proteins produced by B cells in the immune system that help fight off pathogens) ready to attack the virus.
A possible vaccine is of great interest in terms of public health, due to the amount of worldwide infections and the associated health risks from infection by the virus. HCV infects hepatocytes (liver cells) and produces a chronic infection that can result in cirrhosis (scarring) of the liver and cancer of the hepatocytes. Look for more studies to be produced in the near future that follow up on this research and for development of a vaccine that utilizes this new information about the E2 protein.
Article: Khan A, Whidby J, Marcotrigiano J, et al. 2014. Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2. Nature
Michael Houghton, a researcher at the University of Alberta in Canada, has been developing a vaccine that is currently being tested clinically. Houghton, who led a team that discovered the hepatitis C virus in 1989, says the Rutgers finding is important because knowing the structure of the virus will help in the development of a vaccine that enables the immune system to produce more infection-fighting antibodies that can neutralize the virus.
Developing a vaccine against hepatitis C would not only prevent people from acquiring the disease, Marcotrigiano says, but would also be the most cost-conscious health intervention.
-------------------------
Nature | Letter
Nature Feb 19 (2014)
Structure of the core ectodomain of the hepatitis C virus envelope glycoprotein 2
Abdul Ghafoor Khan1, Jillian Whidby1, Matthew T. Miller1, Hannah Scarborough2, Alexandra V. Zatorski1, Alicja Cygan1,
Aryn A. Price2, Samantha A. Yost1, Caitlin D. Bohannon2, Joshy Jacob2, Arash Grakoui2,3 & Joseph Marcotrigiano1
ABSTRACT - Hepatitis C virus (HCV) is a significant public health concern with approximately 160 million people infected worldwide1. HCV infection often results in chronic hepatitis, liver cirrhosis and hepatocellular carcinoma. No vaccine is available and current therapies are effective against some, but not all, genotypes. HCV is an enveloped virus with two surface glycoproteins (E1 and E2). E2 binds to the host cell through interactions with scavenger receptor class B type I (SR-BI) and CD81, and serves as a target for neutralizing antibodies2, 3, 4. Little is known about the molecular mechanism that mediates cell entry and membrane fusion, although E2 is predicted to be a class II viral fusion protein. Here we describe the structure of the E2 core domain in complex with an antigen-binding fragment (Fab) at 2.4 Å resolution. The E2 core has a compact, globular domain structure, consisting mostly of ß-strands and random coil with two small α-helices. The strands are arranged in two, perpendicular sheets (A and B), which are held together by an extensive hydrophobic core and disulphide bonds. Sheet A has an IgG-like fold that is commonly found in viral and cellular proteins, whereas sheet B represents a novel fold. Solution-based studies demonstrate that the full-length E2 ectodomain has a similar globular architecture and does not undergo significant conformational or oligomeric rearrangements on exposure to low pH. Thus, the IgG-like fold is the only feature that E2 shares with class II membrane fusion proteins. These results provide unprecedented insights into HCV entry and will assist in developing an HCV vaccine and new inhibitors.
HCV envelope glycoprotein 2 (E2) is a type I transmembrane protein with an amino-terminal ectodomain connected to a carboxy-terminal transmembrane helix through an amphipathic, α-helical stem (Fig. 1a)5, 6. E2 is highly modified post-translationally with 9-11 N-linked glycosylation sites and 18 cysteine residues that are conserved across all genotypes. For ease of comparison with other genotypes, we refer to the cysteines and N-linked glycosylation sites as C1 to C18 and N1 to N11, respectively, with residue numbers from the J6 genotype (2a) given in parentheses. Full-length, E2 ectodomain (eE2) (384-656) was produced in N-acetylglucosaminyltransferase I-negative (GnTI-) HEK293T cells by a lentiviral expression system and grown in an adherent cell bioreactor. The resulting eE2 protein is monomeric as determined by non-reducing SDS-polyacrylamide gel electrophoresis (PAGE) and size-exclusion chromatography (Extended Data Fig. 1).
Solution-based studies using limited proteolysis and hydrogen deuterium exchange demonstrated that approximately 80 amino acids on the N terminus (384-463) from hypervariable region (HVR) 1 through to HVR2 are exposed and flexible. This region includes conserved sequences implicated in binding to the cellular receptors (SR-BI and CD81) as well as several epitopes for neutralizing antibodies (Fig. 1 and Extended Data Figs 2 and 3)7, 8, 9, 10, 11. Various N-terminal deletions were produced to minimize regions of disorder while preserving an even number of cysteines, potentially allowing for intramolecular disulphide-bond formation. All constructs were screened for aggregation by non-reducing SDS-PAGE and size-exclusion chromatography. E2 core (456-656) is soluble, monomeric and maintains similar secondary structure content when compared with eE2 as determined by reactivity towards HCV-infected patient sera (Extended Data Fig. 4a, b) and circular dichroism (data not shown). However, in contrast to eE2, CD81 binding affinity and the efficiency of inhibition of HCV cell culture (HCVcc) entry was diminished for the E2 core (Extended Data Fig. 4c-e). This indicates that the N terminus of eE2 is critical for CD81 interaction and probably undergoes a transition from disorder to order on binding. Alternatively, the N-terminal region may also be ordered through interactions with other factors, for example, E1, apolipoproteins, lipids, cellular receptors, or antibodies.
Monoclonal antibodies were generated against recombinant eE2 and crystals of deglycosylated E2 core were produced in complex with a Fab (2A12) and diffracted to 2.4 Å resolution (Fig. 1 and Extended Data Table 1). The complex structure was determined by molecular replacement using a Fab structure followed by iterative rounds of model building and refinement. The E2 core domain has a globular fold, consisting of mostly ß-strands and random coil with two short α-helices, which is consistent with previous spectroscopic studies of eE2 (refs 12, 13). The protein contains two, four-stranded antiparallel ß-sheets (termed sheets A and B), the planes of which are approximately perpendicular to each other. The four strands of the N-terminal ß-sheet (sheet A) are stabilized by two disulphide bonds, between strands 1 and 3 (C7 (510) and C8 (554)) and the N-terminal loop with strand 4 (C5 (496) and C9 (566)). The loop between strands 2 and 3 contains sequences implicated in CD81 binding and is flexible, similar to the N-terminal CD81 binding sites, which were deleted14, 15. After strand 4, the polypeptide continues into a long, disordered loop before forming the first short helix (H1) followed by the second ß-sheet (sheet B). A second short α-helix (H2) is located between strands 6 and 7. A disulphide bond (C14 (611) and C16 (648)) between strand 6 and the C-terminal strand 8 further stabilizes the fold. The C-terminal strands (7 and 8) are the longest within the protein with approximately nine amino acids each and encompass the 2A12-binding site. 2A12 does not neutralize HCV infection, indicating that the epitope is either buried within the particle or incapable of preventing entry (Extended Data Fig. 4f). The two ß-sheets are held together by (1) two disulphide bonds, connecting the loops before strand 1 and after H2 (C4 (488) with C15 (624)) as well as the loops after strand 4 and before H1 (C10 (571) and C13 (601)), and (2) an extensive hydrophobic core consisting of numerous aromatic residues (Extended Data Fig. 5).
HCV belongs to the genus Hepacivirus of the Flaviviridae family. Other members of the family include the flavivirus and pestivirus genera, which consist of arthropod-borne viruses and important livestock pathogens, respectively16. The flavivirus envelope glycoprotein (E) is a class II fusion protein and HCV E2 was expected to have a similar fold13, 17, 18. All class II fusion proteins have a common elongated structure, consisting of predominantly ß-sheets, and exist as homo- or heterodimers with the membrane-fusion, hydrophobic peptide buried at the dimer interface at neutral pH. On receptor binding and/or exposure to low pH, these proteins undergo self-rearrangement into stable trimers, exposing the fusion peptide and resulting in viral and host membrane fusion. Despite containing a similar extended organization, the recent structure of the pestivirus bovine viral diarrhoea virus (BVDV) E2 glycoprotein does not represent a typical class II fusion protein fold and lacks an apparent fusion peptide, indicating that it is unlikely to be a class II fusion protein19, 20.
Similar to the flavivirus and pestivirus glycoproteins, the HCV E2 core secondary structure consists of predominantly ß-sheets and random coil. However, E2 core is a monomer with a compact globular shape, in contrast to the extended structures reported in other viruses. Solution-based small-angle X-ray scattering (SAXS) was used to correlate the crystallographic core domain structure with fully glycosylated eE2 and various fragments. The ab initio SAXS envelopes of E2 core and eE2 are similar, with approximately the same radius of gyration (Rg) (Fig. 2a, b and Extended Data Table 2). Glycosylation, which is missing in the E2 core crystal structure, represents roughly one-third of the mass and accounts for the unmodelled areas of the envelopes. Notably, neither the Rg nor the elution profiles on size-exclusion chromatography for fully glycosylated eE2 and E2 core changed significantly at pH 5.0 (Extended Data Fig. 6a, b). These results indicate that unlike class II membrane fusion proteins, E2 does not undergo significant structural rearrangements on exposure to low pH.
SAXS was used to investigate the CD81 binding region on the E2 ectodomain. To simplify data interpretation, eE2(ΔHVR1) was used, as HCV lacking HVR1 remains infectious21. The binding site of CD81 was identified by superimposing the SAXS envelopes of eE2(ΔHVR1) alone and in complex with CD81-LEL (Fig. 2c-e). Although CD81-LEL is a dimer in solution (Extended Data Fig. 6c), the extra density in the SAXS envelope is more consistent with monomeric binding; however, a dimer cannot be ruled out.
HCV E2 is modified by N-linked glycosylation, which is necessary for proper folding and immune invasion. E2 from the J6 genotype has 11 glycosylation sites. Four of the glycosylation sites are in the flexible N-terminal region, which were deleted, and seven are in the core domain (N5-N11). The location of N7, N8, N10 and N11 are modelled in the final E2 core structure. All of these glycans are present in loop areas, indicating that these sites are solvent exposed and flexible. Mutagenesis studies in HCVcc have shown that N6, N8 and N10 are integral for virus infectivity. Removal of the N6 site results in improved CD81 binding, whereas N8 and N10 mutations destabilize the protein and cause defective particle production22. Both sheets have one critical glycosylation site: N8 in sheet A and N10 in sheet B. All four of the observed glycosylation sites are on the periphery of the core and are located on a highly basic surface (Fig. 3). The opposite surface is predominantly hydrophobic and highly conserved when compared to the basic surface. Furthermore, the epitope for antibodies (AR1, AR3A and AR3C) that inhibit E1E2 binding to CD81 is located at the interface of the hydrophobic and basic surfaces, including the N7 glycosylation site (Extended Data Fig. 7a). Interestingly, N7 is only 7 residues away from N6, which has a critical effect on CD81 binding. Epitopes for antibodies (that is, AR5) that block E1E2 heterodimerization are also found on the hydrophobic surface, making it highly plausible that this surface is interacting with E1 in the context of the viral particle4.
The precise roles played by E1 and E2 in membrane fusion are not fully understood. It has been predicted that amino acids 262-290 in E1 as well as 416-430, 504-522 and 604-624 in E2 are important for fusion13, 23, 24. In the structure, the potential fusion regions in E2 (504-522 and 604-624) are located in secondary structure elements within the hydrophobic core and therefore unlikely to serve as the fusion peptide. Furthermore, size-exclusion chromatography and SAXS analyses at low pH indicate that E2 does not undergo oligomeric or structural rearrangement. Thus, it seems unlikely that E2 has a direct role in membrane fusion. However, it is possible that E1 alone or the E1E2 heterodimer has a major role in the fusion process.
Structural comparison of the HCV E2 core domain with all known folds in the Protein Data Bank using the Dali server25 identified proteins with IgG-like folds similar to the N-terminal sheet A, none of which is a class II fusion protein, although IgG-like folds are common in these proteins. The server failed to identify any statistically significant structures to sheet B, suggesting a novel fold. During the review process of this manuscript, a structure for HCV E2 from genotype 1a was published26. The core domain of both structures is highly similar with a root mean squared deviation of 0.8 Å for similar carbon α atoms. Our biochemical and structural data provide valuable information towards defining the role of E2 and establish a foundation for further studies in understanding HCV entry and infection.
|
|
| |
| |
|
|
|