| |
Eradicating HIV-1 infection: seeking to clear a persistent pathogen - Review
|
| |
| |
Download the PDF here
"The scientific and medical challenges in the effort to eradicate HIV-1 infection are formidable and complex. Ultimately, given the scope of the HIV-1 pandemic, strategies to eradicate the disease must be implemented globally........The development of approaches to eradicate HIV-1 infection will take time and durable investment in research towards this goal. It is possible that therapies that result in the depletion of persistent infection and the augmentation of the immune response might lead to an intermediate result. Termed by many as a 'functional cure', this is a state in which HIV-1 infection is not cleared but is so tightly controlled by the immune response that the patient is no longer infectious and is clinically stable in the absence of ART. If so, to make such an investment superior to once-a-day ART (or less with the long-acting therapeutic agents that are currently under development), interventions that result in a functional cure would also have to spare patients the chronic immune activation that is seen in natural 'elite controllers' of HIV-1 infection173, with its attendant risks of long-term morbidity, as otherwise life-long ART might then be clinically preferable......
......As efforts advance, additional obstacles to clear HIV-1 infection are likely to be uncovered, and careful consideration must be given to the ethics of translational research with otherwise healthy volunteers infected with HIV-1. The recent reinvigoration of efforts to gain a detailed understanding of the biology and pathogenesis of viral latency should give hope that we can overcome these obstacles. The journey towards a cure for HIV/AIDS has begun.......Efforts to develop therapies that could eradicate HIV-1 infection or achieve a durable remission of viraemia in the absence of ART have recently accelerated and expanded. Although this initial period of renewed effort has been marked by much progress and enthusiasm, both the scientific and the patient community must be prepared for the prolonged effort that will be required to overcome both the expected and the unforeseen challenges ahead. First, and perhaps most daunting, is the need to target latency within specific cellular reservoirs to disrupt viral quiescence so that residual infection can be cleared.......The several approaches to disrupt HIV-1 latency by inducing proviral expression seem to be promising and might be implemented in the foreseeable future. As reactivation of the latent reservoir may be governed by stochastic mechanisms (that is, some latent genomes remain silent even in the event of a single round of maximal mitogen stimulation)138, combinatorial latency-reversing therapy that is safe enough to enable multiple administrations may be needed. Ongoing work aims at designing an effective dosing regimen. Potency must be balanced with minimizing toxicity, and the potential impact of any latency-reversing agent on the immune system must be carefully considered........In addition, a parallel effort must be made to develop immune-based therapies to ensure and accelerate the process of viral clearance."
-----------------------
Eradicating HIV-1 infection: seeking to clear a persistent pathogen
Nature Reviews Microbiology
16 October 2014
Nancie M. Archin1, Julia Marsh Sung1, Carolina Garrido1, Natalia
Soriano-Sarabia1 and David M. Margolis1-3
1Department of Medicine, University of North Carolina at Chapel Hill.
2Department of Microbiology and Immunology, University of North Carolina at Chapel Hill.
3Department of Epidemiology, University of North Carolina at Chapel Hill, Chapel Hill, North Carolina 27599, USA.
Abstract
Effective antiretroviral therapy (ART) blunts viraemia, which enables HIV-1-infected individuals to control infection and live long, productive lives. However, HIV-1 infection remains incurable owing to the persistence of a viral reservoir that harbours integrated provirus within host cellular DNA. This latent infection is unaffected by ART and hidden from the immune system. Recent studies have focused on the development of therapies to disrupt latency. These efforts unmasked residual viral genomes and highlighted the need to enable the clearance of latently infected cells, perhaps via old and new strategies that improve the HIV-1-specific immune response. In this Review, we explore new approaches to eradicate established HIV-1 infection and avoid the burden of lifelong ART.
Introduction
HIV-1 infection remains incurable owing to the presence of quiescent, replication-competent provirus within a long-lived population of memory T cells, which are capable of reigniting new rounds of infection if therapy is interrupted. In adults, this latent pool of virus is established within days of infection and is unaffected by the antiviral immune response or by current therapy. HIV-1 preferentially infects activated CD4+ T cells , which leads to massive depletion of these cells, as well as the accompanying immune suppression and exhaustion that are characteristic of HIV-1 infection. Infection begins when the HIV-1 envelope (Env) engages the CD4 receptor and a CC-chemokine receptor, usually CCR5 and rarely CXC-chemokine receptor 4 (CXCR4), on the surface of host cells, which leads to fusion of the viral and cellular membranes and thus enables entry of the viral nucleocapsid into the cell. The viral RNA genome is reverse transcribed into proviral double-stranded cDNA, which together with viral and host cellular proteins forms the pre-integration complex (PIC). This complex is imported into the nucleus, where integration of the proviral cDNA into the host genome occurs. In activated T cells, infection proceeds with the transcription of viral mRNAs, protein production and the generation of new viral particles. In resting T cells, the provirus may enter quiescence, whereby it exists in a latent state as part of the host gene in which it is integrated.
Several classes of drugs that target the different stages of the viral life cycle have been successfully used in combination antiretroviral therapy (cART) for the treatment of HIV-1 infection. These include: fusion inhibitors and CCR5 co-receptor antagonists, which block viral entry; nucleoside reverse transcriptase inhibitors (NRTIs) and non-nucleoside reverse transcriptase inhibitors (NNRTIs), which block reverse transcription of the viral genome; integrase inhibitors, which prevent viral integration; and protease inhibitors, which interfere with virion production. However, there are currently no available therapies that target the quiescent integrated form of the virus, and unless this persistent latent infection is eradicated, HIV-1 will remain a chronic viral infection with the enduring potential to cause or spread lethal disease.
Although disappointing, the recent return of viraemia in an infant born to an HIV-1-positive mother (known as the 'Mississippi baby')1 more than 2 years after the interruption of ART suggests that individual latently infected cells may remain dormant for considerable periods of time, and perhaps, if the number of latently infected cells is low enough, an antiviral immune response may stringently contain infection. HIV-1 rebounded only several months after stopping treatment in two patients (known as the 'Boston patients') who received bone marrow transplants to treat lymphoma2. The shorter time off therapy before rebound in the Boston patients might simply reflect a higher number of latently infected cells in the adult patients and/or the absence of memory T cells that could harbour quiescent, replication-competent provirus in the Mississippi baby at birth. Approaches to disrupt latency or durably enforce latency, in combination with effective therapeutic agents that continuously enhance the immune response to HIV-1 infection, must now be even more seriously considered.
In this Review, we briefly describe the main mechanisms that are involved in the establishment and maintenance of HIV-1 latency and discuss cellular HIV-1 reservoirs, including memory T cells and their precursor cells, as well as myeloid cells, with a focus on macrophages. We then discuss the current cell and animal models that are available for the study of HIV-1 latency and the proposed strategies to disrupt latent infection and enable clearance of persistently infected cells.
HIV-1 latency
Latently infected resting memory CD4+ T cells are the best characterized reservoirs of HIV-1 infection. These are a small population of cells that, rather than dying from the direct or indirect cytopathic effects that are induced by the virus, persist after infection as long-lived cells that harbour integrated HIV-1 DNA in their genomes3. This latent reservoir is established within days of acute infection4, with continued contributions from active, uncontrolled viraemia in the absence of ART, and although early treatment with ART can reduce the size of this pool of infected cells, it cannot prevent the establishment of latent, persistent HIV-1 infection5, 6.
Infection of resting CD4+ T cells is far less efficient than infection of activated cells7, 8, 9, 10, which express factors that are crucial for HIV-1 transcription. Latency may primarily be established in activated CD4+ T cells that are infected as they transition to the resting memory state11. However, recent studies using primary CD4+ T cells that are infected with dual-labelled HIV-1 reporter viruses suggest that a small fraction of transcriptionally silent infection occurs directly in activated CD4+ T cells that have not yet transitioned to a resting state12. Whether this phenomenon occurs in vivo remains to be determined. HIV-1 persists in resting cells, but is transcriptionally silent and therefore 'hidden' from immune surveillance and unaffected by ART4, 13. However, as latent HIV-1 still remains replication-competent and has the ability to re-emerge when therapy is interrupted, it poses a considerable barrier to the eradication of HIV-1. Furthermore, there are currently no known cellular biomarkers that distinguish latently infected cells from uninfected cells, although it has been shown that some latently infected resting CD4+ T cells express high levels of CD2 (Ref. 14).
Despite some evidence to the contrary15, 16, ongoing viral replication has been suggested to contribute to the persistence of HIV-1 infection even in the presence of therapy. Sensitive assays have detected trace levels of viraemia in many ART-treated patients17, 18. This phenomenon seems to be the result of continuing viral expression from cells that were infected before the implementation of ART. However, it has been suggested that ongoing viral replication in a pro-inflammatory environment within lymphoid tissue contributes to the maintenance of persistent infection. Evidence for ongoing viral replication has previously been reported19, 20, 21 and cell-to-cell spread of HIV-1 was recently proposed as a mechanism that facilitates ongoing replication despite ART22; however, these findings have been challenged23, 24, 25.
The adequacy of ART is an area of continued controversy. Treatment intensification with the integrase inhibitor raltegravir had no effect on low-level viraemia, but it was associated with an increase in the generation of circular HIV-1 DNA episomes that contain two copies of the two-long terminal repeat circles (2-LTR circles)21, 26 and a reduction in the levels of recoverable HIV-1 (Ref. 27). However, these findings were not seen in all raltegravir intensification studies28.
Another recent study observed persistent HIV-1 RNA expression in tissues (although after only 6 months of therapy) and low levels of some antiretroviral drugs in some tissues29. However, it has been noted that, although intracellular active nucleotide metabolites remain stably inside cells during processing30, parent drugs quickly diffuse out of cells31, which makes it difficult to obtain accurate measurements of active drug levels. The amount of NNRTIs, protease inhibitors or integrase strand transfer inhibitors (INSTIs) that are lost from cells during isolation is currently unknown. Furthermore, the fact that low-level plasma viraemia remained unaffected following the intensification of therapy and the lack of genetic evolution of plasma virus20, 32, 33, 34, 16 leaves many in this field of research convinced that residual viral replication may not be involved in the maintenance of persistent HIV-1 infection.
Homeostatic proliferation of latently infected cells may alternatively, or additionally, contribute to the maintenance of this cell pool35, 36. Recent studies have shown an enrichment of HIV-1 DNA integrated in, or near to, host genes that are associated with cell cycle control. These findings suggest an alternative mechanism for proviral persistence, whereby the integration of HIV-1 into such sites could lead to proliferation of the latently infected cells37, 38. However, further experiments are necessary to fully examine this theory, and it is possible that the observed results were reflective of accumulated, defective proviral DNA rather than truly replication-competent virus39. Like host gene expression, the latency of integrated, proviral DNA is regulated by multiple cellular mechanisms, including epigenetic transcriptional silencing, the availability or deficiency of key host factors and transcriptional interference (reviewed in detail in Ref. 38). Briefly, host transcription factors, including nuclear factor-κB (NF-κB), nuclear factor of activated T cells (NFAT), AP1 and SP1, are sequestered in the cytoplasm in resting cells and thus do not promote HIV-1 transcription until an appropriate cellular activation signal is transmitted (Fig. 1). HIV-1 integration into the host genome preferentially occurs within introns of actively transcribed host genes40, 41, 42. Multiple distinct and complementary mechanisms contribute to the establishment of latent proviral infection (Fig. 1).
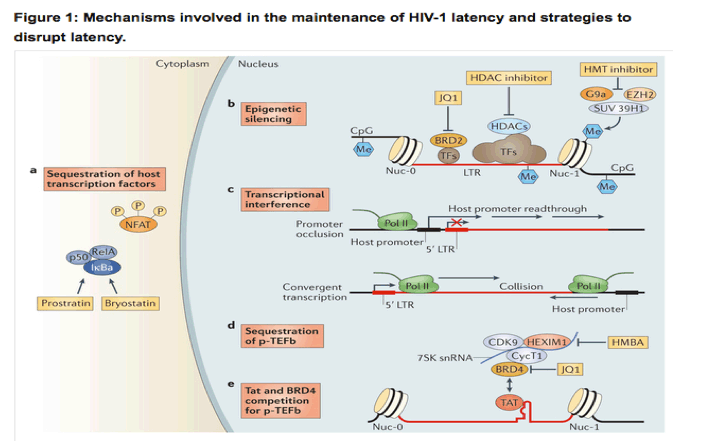
HIV-1 latency is maintained by several mechanisms. a | Transcription factors (TFs), including nuclear factor-κB (NF-κB) and nuclear factor of activated T cells (NFAT), are sequestered in the cytoplasm, which leads to transcriptional silencing. Bryostatin and prostratin induce activation of NF-κB, leading to its translocation to the nucleus where it activates HIV-1 transcription. b | The HIV-1 long terminal repeat (LTR) is flanked by the Nuc-0 and Nuc-1 nucleosomes that, when latent, can encode repressive post-translational histone modifications. Histone deacetylases (HDACs), which are recruited by transcription factors (such as YY1 and CBF-1), remove the acetyl groups from chromatin. Histone methyltransferases (HMTs), such as SUV39H1, G9a and EZH2, deposit methyl groups onto histones. HDACs and HMTs enforce the repressive state. Both HDAC inhibitors and HMT inhibitors can induce transcription from quiescent LTR promoters. HIV-1 DNA can also be methylated, although recent evidence suggests that DNA methylation is an epiphenomenon that does not play a part in HIV-1 latency. Bromodomain-containing (BRD) proteins have a complex role in HIV-1 transcription initiation and processivity. Recent evidence suggests that BRD2 has a unique role in enforcing HIV-1 latency, and therefore, BRD inhibitors such as JQ1 may be of use as latency-reversing agents. c | Transcriptional interference may contribute to the regulation of HIV-1 latency. If viral DNA is integrated within an intron of an upstream host gene, readthrough of RNA polymerase II (Pol II) displaces key transcription factors on the HIV-1 LTR (known as promoter occlusion). Conversely, if the viral genome is integrated in the opposite polarity relative to the host gene, host RNA Pol II complexes may induce premature termination of HIV-1 transcription (known as convergent transcription). d,e | The positive transcription elongation factor b (p-TEFb) complex (which comprises CDK9 and cyclin T1 (CycT1)) is sequestered in an inactive ribonucleoprotein complex with HEXIM1-7SK small nuclear RNA (snRNA). BRD4 may compete with the viral Tat activator for binding to p-TEFb. Hexamethylene bisacetamide (HMBA) releases p-TEFb from the HEXIM1-7SK snRNA inhibitory complex and the small-molecule inhibitor JQ1 may antagonize BRD4, both of which enable induction of latent HIV-1 expression.
-----------
Epigenetic modifications are involved in the initial establishment and subsequent enforcement of transcriptional silencing of the provirus. Independent of the site of integration, the viral 5' LTR is occupied by two specific nucleosomes - Nuc-0 and Nuc-1 (Ref. 43) - that can be marked by repressive post-translational histone modifications44, 45. Histone deacetylation is associated with transcriptional repression of the HIV-1 promoter, and inhibition of histone deacetylases (HDACs) reactivates latent HIV-1 (Ref. 46). Histone methyltransferases (HMTs), such as EZH2, G9a and SUV39H1, have also been suggested to contribute to latency in T cells45, 47. These chromatin marks create an environment that favours the recruitment of additional factors and complexes that antagonize proviral gene expression.
An additional, or complementary, mechanism to suppress the expression of the integrated provirus is transcriptional interference, which involves the integration of the provirus in the same orientation but downstream of an actively transcribed host gene (known as promoter occlusion) or the integration provirus in the opposite orientation relative to the host gene (known as convergent transcription) (Fig. 1). Transcriptional elongation is suppressed by sequestration of positive transcription elongation factor b (p-TEFb), which comprises cyclin-dependent kinase 9 (CDK9) and cyclin T1 (Refs 48, 49) and associates with the HEXIM1-7SK small nuclear RNA (snRNA) regulatory complex. CDK9 is constitutively expressed in resting cells in an inactive dephosphorylated form50. HIV-1 Tat recruits p-TEFb to the viral promoter, where active CDK9 can promote transcriptional elongation46 (Fig. 1).
Finally, microRNAs (miRNAs) may contribute to latency, although their specific role has not yet been fully established (reviewed in Ref. 51). The factors and steps that lead to complete HIV-1 transcription after reactivation have recently been reviewed46.
Cellular reservoirs of HIV-1
It is crucial to identify and fully characterize all reservoirs of persistent HIV-1 infection so that specific therapies can be devised. The nature of these reservoirs is still a subject of controversy. To meet the criteria of a long-lived, latent reservoir of HIV-1 infection, an infected cell population must persist for months, restrict viral expression to the extent that viral antigen is not presented and harbour quiescent virus that is replication-competent following reactivation.
Memory CD4+ T cells. HIV-1 DNA is primarily detected in two subsets of memory CD4+ T cells: central memory CD4+ T cells (TCM cells) and transitional memory CD4+ T cells(TTM cells)35. TTM cells are characterized by the expression of CD27 but lack expression of the lymph node homing receptor CCR7, whereas TCM CD4+ T cells express both CD27 and CCR7. The frequency of infection of TCM cells is associated with the presence of human leukocyte antigen-B27 (HLA-B27) and HLA-B57, which have been shown to have a protective role in long-term non-progressors52.
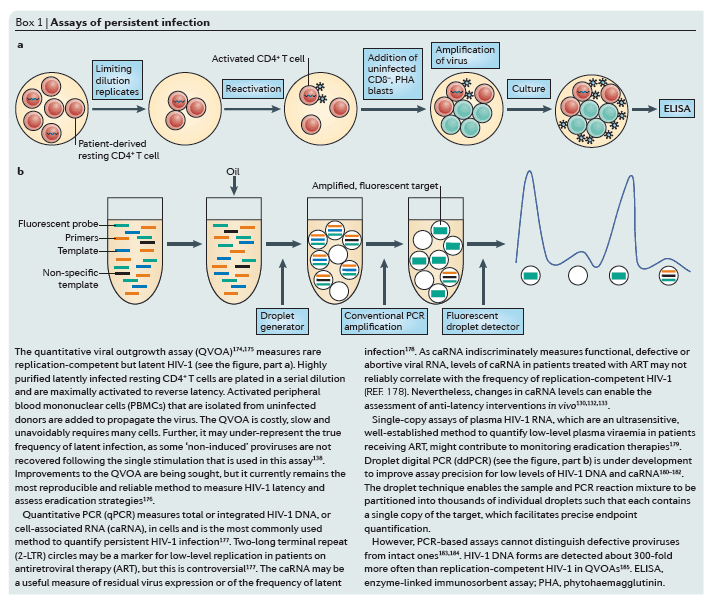
Interleukin-7 (IL-7)-mediated homeostatic proliferation was reported to be the main mechanism that maintains latency in TTM cells in patients with low CD4 counts, whereas antigen-driven T cell receptor (TCR) activation maintains the reservoir in TCM cells35, although in this case the hypothesis holds that antigen-driven activation must not be sufficiently strong to induce reactivation of HIV-1 from these cells36. Another recent study found that HIV-1 DNA and RNA levels were high among TTM cells in patients who are early in the course of infection53. However, preliminary analysis of the frequency of replication-competent virus by a quantitative viral outgrowth assay (QVOA) (Box 1) in TCM and TTM cells showed that TTM cells are major contributors to the HIV-1 reservoir in only a minority of the patients studied, and in those patients, infection of TTM cells does not seem to persist as it does in TCM cells54, 187. Owing to the challenges that are inherent in comparing measurements of rare events in small populations of cells, further longitudinal analyses are needed to clarify whether TTM cells constitute a durable and clinically significant reservoir in patients who are successfully treated with ART and who have normal CD4 counts.
Other T cell subsets. In addition to the well-established memory CD4+ T cell reservoir, it is possible that less differentiated populations of CD4+ T cells function as long-term, cellular reservoirs. The role of CD34+ haematopoietic progenitors as additional reservoirs for HIV-1 has been controversial for many years55; for example, HIV-1 infection and the establishment of latent infection in CD34+ haematopoietic progenitor cells in patients on ART have been reported56, 57, but these studies were not confirmed by others58, 59. If progenitor cells are durably latently infected, they could be a source of persistent HIV-1 production when these cells go through proliferative cycles. However, the initial description of HIV-1 infection in these haematopoietic stem and progenitor cells found that proliferation resulted in the death of these infected cells in vitro56, 57. Therefore, although this reservoir might be established before therapy, it would be expected to decay over time during treatment.
In addition to memory CD4+ T cells, latent infection of naive CD4+ T cells in patients on ART has also been shown, although this event seems to occur with even lower frequency35, 60. A recent study reported that CD4+ memory stem cells (TSCM cells) constitute a novel HIV-1 reservoir that may not be large but that seems to be stable over time61. TSCM cells are a population with characteristics of stem cells: they have a less differentiated phenotype and are reported to be extremely long-lived62, 63. The contribution of these TSCM cells to the total HIV-1 reservoir, which is reported to be less than 10%, may be especially relevant in patients with small TCM cell reservoirs.
Preliminary work also detected the presence of replication-competent virus and HIV-1 DNA in γδ T cells, which are a subset of CD3+ T lymphocytes that harbour alternative TCRs formed by γ-chains and δ-chains54. The biology of γδ T cells differs from conventional αß T cells; for example, γδ T cells do not recognize peptide antigens but rather recognize lipid antigens in a major histocompatibility complex (MHC)-unrestricted manner64, 65, 66, 67, 68. These differences in the signalling and activation of γδ T cells suggests the induction of latent HIV-1 provirus in γδ T cells may have different requirements.
Persistent infection of macrophages. Enduring infection of macrophages, their precursors or other myeloid lineage cells, such as dendritic cells, has long been a concern. Infection of macrophages, with the potential for both high-level viral production in the presence of macrophage-derived pro-inflammatory cytokines, and the potential for durable viral production, given the resistance of macrophages to viral cytopathicity in vitro, was first demonstrated nearly 30 years ago69, 70, 71. HIV-1 has been recovered from the circulating monocyte pool of patients treated with ART72, but the durability of this reservoir has not been carefully measured as it has in memory T cells. Brain astrocytes, microglia and other related cellular lineages in the brain have been shown to support a restricted infection that could persist despite ART73, 74. However, even after years of study, a clear demonstration that macrophages and these other related cells are truly latent viral reservoirs is lacking. Such cells have not fulfilled the strict definition of latency, which is the recovery of cells from a patient or animal on durable, suppressive ART that produce virions only following activation.
However, although definitive proof is lacking, several characteristics of these cell types would seem to make them ideal reservoirs for long-term infection. The long-standing view of macrophages as terminal cells in the myeloid differentiation pathway has recently changed, and the ability of macrophages to self-renew and repopulate tissues has been appreciated75. This presents the possibility that, like T cells, infected macrophage populations could persist despite low-level virion production and clearance of some infected cells, if at least one-half of the dividing macrophages escaped viral cytolysis. It is crucial to be clear about the difference in persistence of HIV-1 infection in myeloid cells - in which cells may survive for long periods of time while viral genomes are in a state of non-latency and low-level HIV-1 expression - and true virological latency in CD4+ T cells - in which the viral genomes must be mostly silenced, without any appreciable expression of viral proteins. This distinction results in key differences in methods to detect persistent infection in myeloid cells and to eliminate persistent infection (Box 1); for example, the challenge of disrupting latent infection in CD4+ T cells may be of little relevance in macrophages if productive infection already persists owing to very low-level viral expression. If this is the case, we would instead need to develop interventions to assist in the clearance of persistently infected myeloid cells.
Recent advances in the implementation of fully effective ART in the non-human primate (NHP) SIV model system (see below) offer hope that this controversy will soon be resolved76, as extensive and conclusive tissue sampling of optimally treated animals should be possible. In addition, recently developed humanized mouse models of latent HIV-1 infection will address this question in the context of the human virus in human cells77. The flexibility of the humanized BLT (bone marrow-liver-thymus) mouse has recently enabled the generation of animals that have macrophages but that do not have T cells78. If infection persists in this model system in the presence of continuous therapy, this would provide definitive proof of a latent reservoir in macrophages.
Model systems of HIV-1 latency
No model can fully recapitulate the complexities of the latent reservoir in vivo. However, as clinical trials will always necessarily limit therapeutic interventions to those with reasonable expectations of safety and efficacy, models will continue to have a crucial role in HIV-1 eradication research (Fig. 2).
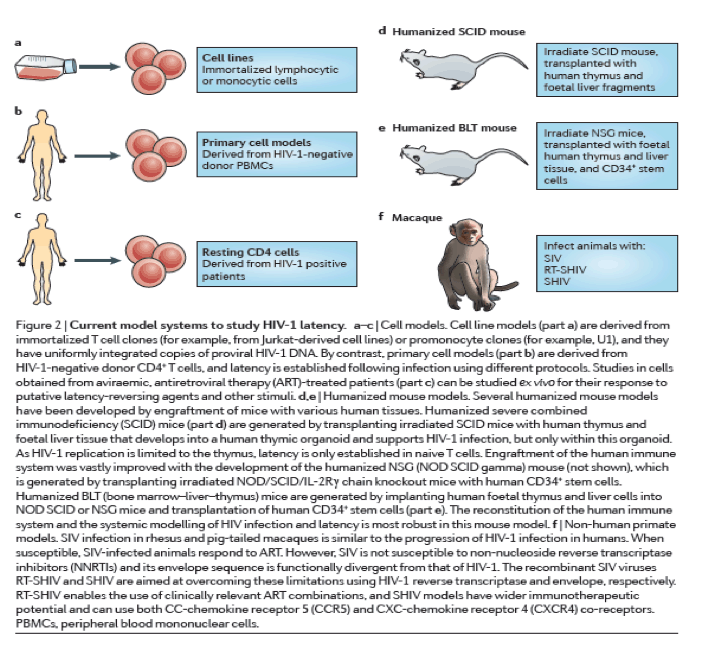
Cell models of latency. Cell models have been particularly useful for understanding the basic mechanisms that are involved in establishing and maintaining latency as well as for the initial screening of latency-reactivating agents79 (Fig. 2a-c). Although resting memory CD4+ T cells constitute the major latent HIV-1 reservoir in humans, studies using these cells are limited by the very low frequency of infection80. Primary cell models aim to overcome these limitations by establishing latent HIV-1 infection in CD4+ T cells that have been isolated from HIV-1-negative donors at a higher frequency. As such cells are not immortalized or clonal, primary cell models might offer a more physiologically relevant representation of latency than cell lines, and their responsiveness to stimuli might be more representative of in vivo biology. Several cell models are currently being used81, 82, 83, 84, 85, 86, 87, 88 (reviewed in Refs 89, 90), and there are important differences in the cell subsets, viral strains and mechanisms that are used to establish latency89, 90 (Table 1). A careful comparison of latent HIV-1 reactivation by different stimuli across multiple primary cell models revealed diverse responses to the same stimuli. In addition, such responses are not uniformly consistent with results obtained using QVOA in resting CD4+ T cells that have been isolated from aviraemic patients infected with HIV-1 (Ref. 89) (Box 1). For example, HIV-1 expression can be induced in J-lat clones and primary cells from patients by HDAC inhibitors, whereas several primary cell models of latency are resistant to the effects of HDAC inhibitors. Given the complexity and diversity of latency in vivo and the varying responses of these models, information from primary cell models may be expanded by evaluating responses in more than one model91.
Humanized mouse models. The establishment of successful mouse models of latent HIV-1 infection required substantial adaptation of the host (Fig. 2d,e), as mice are not naturally susceptible to HIV-1 infection (reviewed in Ref. 92). The development of the humanized BLT mouse77, 93, 94, 95, 96, 97, which is characterized by a complete reconstitution of human immune cells, including full mucosal immunity, has greatly increased the ability to study the distribution of infection and latency77, 96, 98, 99, 100, 101, 102 (Fig. 2e). A recent study102 extensively characterized sites of residual active viraemia during ART in blood and multiple tissues and found reduced, but detectable, residual viral RNA expression in all tissues (especially lymphoid tissues), despite adequate tissue drug concentrations. The humanized BLT mouse model also enables manipulation of the immune system to generate HIV-1-resistant cells and/or an enhanced immune response via transgenic or short hairpin-mediated modifications of CD34+ stem cells before transplantation103, 104. Further adaptations of humanized mice that are aimed at isolating discrete cell populations of interest are currently being developed; for example, using a novel T cell-only humanized mouse model105, a recent study showed that latency is established in resting CD4+ T cells despite the absence of monocytes, macrophages, B cells and dendritic cells.
NHP models. Animal models are well-suited for the study of the anatomical and cellular distribution of latent infection in the setting of ART treatment, as well as for the evaluation of certain high-risk treatments that would be unethical for initial human trials (Fig. 2f). SIV and recombinant viral strains derived from SIV (such as RT-SHIV and SHIV) cause a pathogenic disease course in Asian macaques that is similar to that of HIV-1 in humans, and treatment with ART results in plasma viral decay106, 107, 108, 109, 110 and the establishment of inducible replication-competent virus in resting CD4+ T cells106, 111. Neurotropic strains of SIV, such as SIV/17E-Fr, have also been used to establish models of central nervous system (CNS) disease during infection and ART107, 112, 113, 114. These studies have shown the establishment of HIV-1 DNA early in the course of infection, despite the initiation of ART during acute infection, although the specific cellular reservoirs within the brain tissue were not delineated and studies were limited by reliance on evaluation of HIV-1 DNA rather than replication-competent virus. The development and application of recombinant RT-SHIV115 has provided further approaches to characterize latent reservoirs following treatment with clinically relevant ART regimens116, 117. RT-SHIV-infected macaques that were treated with ART were found to have a widespread distribution of both viral RNA and DNA, especially in the gut and lymphoid tissues116.
The SIV-macaque model was also key to understanding the role of the immune response in controlling HIV-1 infection118. Recent studies using a rhesus cytomegalovirus (CMV) vector vaccine in macaques, followed by infection with SIV, led to suppression of viraemia below the limit of detection in 50% of animals119, 120. Although correlates and mechanisms of protection are still under investigation, interestingly, the rhesus CMV vector induced a non-canonical, MHC II-restricted CD8+ T cell response. It is unclear how directly translatable the magnitude of the results will be in human trials, especially for patients who started ART during chronic therapy. Autologous pre-infected CD34+ haematopoietic stem cell (HSC) transplants have also been used in SIV-infected macaques as a tool to investigate the potential contribution of non-haematopoietic reservoirs to the persistence of HIV-1 (Ref. 121). In addition, transplantation of autologous HSCs that were genetically engineered to be HIV-1-resistant has also been explored as a therapeutic strategy in macaques122. However, although similar to humans, the macaque immune response to SIV is distinct, and results that are obtained in macaques might not translate to the human system.
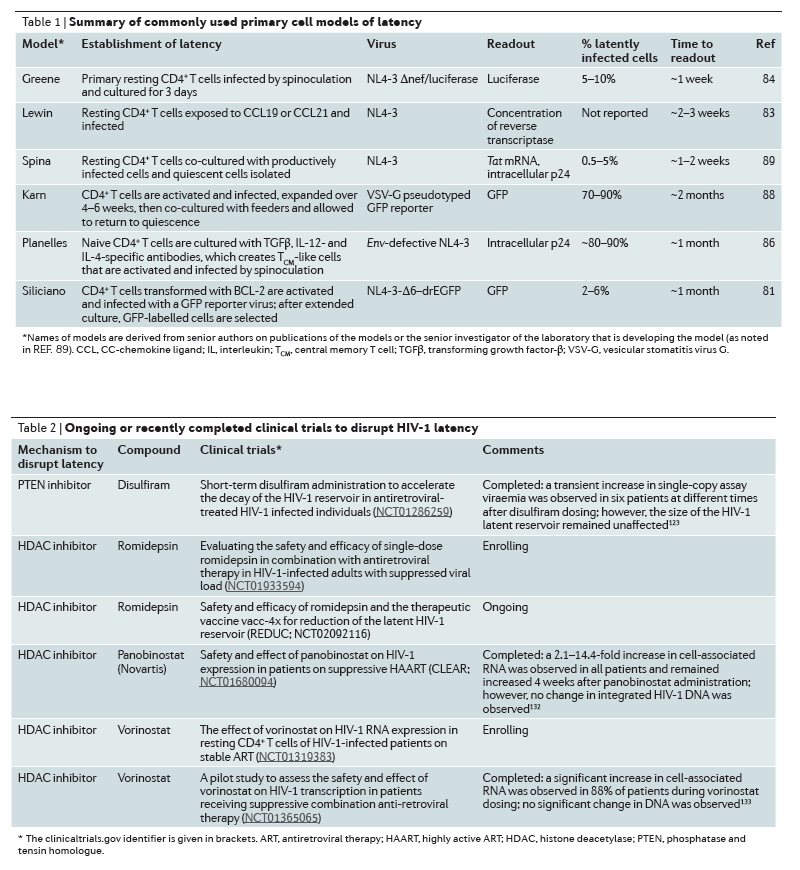
Strategies to disrupt latent infection
Several modalities to target the latent reservoir have been proposed. These interventions aim at inducing proviral expression to enable clearance of the virus and infected cells123 (Fig. 1; Table 2). Early attempts to reactivate virus production via global T cell activation using OKT3 and IL-2 in combination led to toxic levels of immune activation, and thus current strategies focus on reactivating the virus in the absence of T cell activation124. However, most of these approaches have been validated only in cell line models of HIV-1 latency, and only a few have been tested in resting CD4+ T cells that have been isolated from aviraemic patients51, 91, 125, 126, 127, 128.
Many primary cell models of latency have recently been developed, and the responses of these models to a panel of reagents that are known to induce LTR expression were compared in a comprehensive study89. Although some of these models reflect the responses that have been obtained in resting CD4+ T cells that have been isolated from aviraemic patients, none precisely reflects the responses of cells from patients to every type of anti-latency modality that has been tested89. Furthermore, a recent report using resting CD4+ T cells from aviraemic patients showed that latency-reversing agents, such as HDAC inhibitors, only weakly induced HIV-1 transcription129, but it is difficult to directly compare the precise assays and conditions that were used in this study to those measured in a clinical trial130. Therefore, in primary polyclonal cell models of HIV-1 latency, cell-specific factors present a challenge to the use of ex vivo cell systems for the validation of potential anti-latency approaches, and even the research assays that are used to evaluate cells from patients still lack a relevant clinical endpoint (that is, depletion of latent infection) to validate the relevance of the effects measured.
Nevertheless, the use of small-molecule inhibitors that target HDACs and induce transcription at the HIV-1 LTR remains the most well-characterized strategy to purge latent HIV-1 (Ref. 131), and several HDAC inhibitors have advanced into clinical trials132, 133, 134 (Table 2). A single 400 mg dose of the class I HDAC inhibitor vorinostat, can disrupt latency in humans, as measured by the expression of HIV-1 RNA in isolated resting CD4+ T cells130. The finding of increased levels of HIV-1 RNA transcription on the first day of therapy has been replicated in a study in which 14 daily doses of vorinostat were administered133 and in another study with the HDAC inhibitor, panobinostat, which was administered three times a week for 4 weeks with weekly off-drug intervals132. These studies measured HIV-1 RNA in total CD4+ T cells rather than in isolated resting CD4+ T cells. In addition, these studies found that, compared with a single baseline pre-dose measurement, the levels of unspliced HIV-1 RNA within total CD4+ T cells is increased throughout the dosing period and is still increased 84 days after the initial dose of the HDAC inhibitor.
However, a subsequent follow-up study observed a dampened response to vorinostat following administration of multiple daily doses to patients who responded to the single dose135. Preliminary gene expression analysis of resting CD4+ T cells treated ex vivo with a single dose of vorinostat shows a complex multiphasic cascade of host gene expression (D.M.M., unpublished observations). Furthermore, dose-response measurements in patients who received multiple doses of vorinostat showed an exposure-effect relationship with clockwise hysteresis (that is, the response to the initial dose was higher than the response to the subsequent doses), which is consistent with tolerance to vorinostat exposure136. Taken together, these results suggest that further understanding of the kinetics of the effects of vorinostat on a crucial subset of host genes - perhaps those that maintain repression of HIV-1 transcription - may be necessary to design a dosing regimen that can lead to effective and durable induction of latent HIV-1 genome expression.
Reactivation of HIV-1 expression by itself may not lead to reservoir clearance, and whether virion production is necessary to achieve viral clearance has not been proven. It is plausible that any viral antigen that is expressed by a latently infected cell that has been stimulated may be sufficient for natural killer cell- or cytotoxic T lymphocyte (CTL)-mediated clearance137. Clinical trials to test this hypothesis are in the planning stage.
Successful strategies to disrupt latency are likely to include cycles of combination therapy that target distinct mechanisms that maintain latency. As shown in recent work138, about 10% of integrated provirus that does not express detectable HIV-1 RNA following a single round of maximal mitogen stimulation may still be fully replication competent, which suggests that more than a single round of in vivo T cell stimulation will be required to purge the reservoir.
A combinatorial effect of drugs that inhibit HDACs or HMTs, or that induce protein kinase C isoforms (to induce NF-κB-mediated LTR transcription), have been described in various transformed cell line systems, but how these results will be successfully translated to implementation in vivo is unclear139, 140, 141, 142. Synergistic combination drug therapy to target latent provirus is difficult to define and measure. Synergy of multiple drugs that target different mechanisms of latency might induce the expression of a greater proportion of latent proviruses or induce expression to a greater extent, which might lead to the death of infected cells or improve the recognition and clearance of infected cells by the immune system. However, the level of proviral expression that will result in cell death or in immune recognition and clearance has yet to be determined. It seems likely that a highly potent induction of the latent virus could result in host toxicity and/or levels of viral expression that could not be contained by ongoing ART. Synergy has often been described as a combined effect that is greater than the sum of the effect of two separate modalities. However, a recent study cautioned against this simplistic definition of synergy, given the complexity of biological systems, and suggested the use of the Chou-Talalay method143 to more accurately measure the effects of multiple antiretroviral drug therapy.
cART suppresses plasma viraemia and controls HIV-1 infection by targeting specific viral enzymes and inhibiting fusion and entry, which enabled the development of well-established laboratory models that predict clinical effects. Combination cancer chemotherapy and immunotherapy have only recently been successfully used after many years of intense research. Lessons from both fields may offer insights into how to proceed with combinatorial latency eradication approaches.
An important area for future study is to establish validated cell and animal model systems that can reliably evaluate combinatorial approaches to disrupt HIV-1 latency. An initial study in the humanized mouse model that examined the effect of a novel immunotoxin that recognized and killed cells expressing HIV-1 Env when added to an antiretroviral drug regimen has recently been reported102, but the impact of interventions on latent persistent infection has not yet been successfully tested in this model or in NHPs. Many questions arise in such investigations, such as: which response parameters are predictive of disruption of latency in vivo; what is the temporal manner in which reagents are delivered, in series or in parallel, in what order and for what duration; do these reagents access all the relevant tissue compartments in which latently infected cells reside; do these reagents induce clearance without other interventions; if interventions work via a host cell response (for example, vaccines, antibodies or cytokines; see below), do host-targeted anti-latency therapies affect such adjunctive therapies?
Clearing persistently infected cells
The induction of latent proviral expression may not be sufficient to clear latently infected cells by viral cytopathic effects alone144, but the expression of HIV-1 antigens may enable the immune system to identify latently infected cells. However, continuous antigenic stimulation during HIV-1 infection leads to chronic immune activation and immune exhaustion, and therefore HIV-1-specific effector cells are depleted or dysfunctional as they lose antiviral function and proliferative capacity145. An effective eradication strategy is likely to require interventions to improve the HIV-1-specific immune response (Fig. 3).
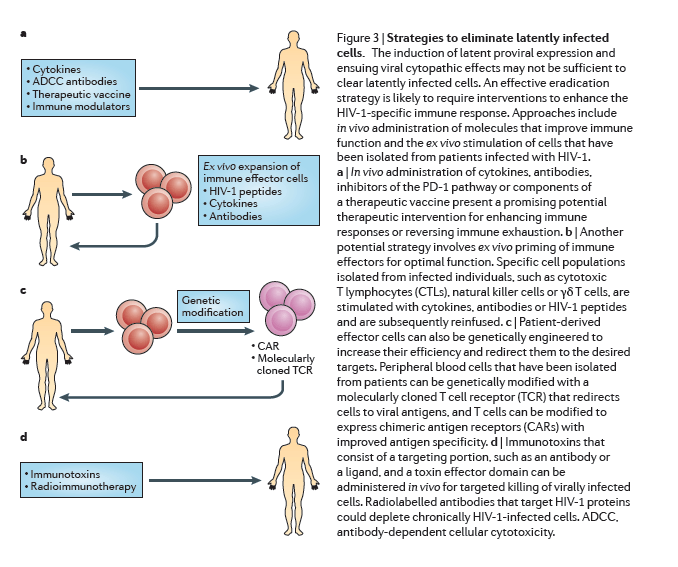
Therapeutic vaccines. HIV-1 infection compromises T cell effector function and also provokes B cell exhaustion, which may result in an inadequate antibody response146. The objective of a therapeutic vaccine is to enhance the immune response against infection using a controlled in vivo exposure to HIV-1 antigens. The rebound viraemia that was observed in the Mississippi baby after 27 months of ART interruption poignantly illustrates the need for a robust, durable antiviral immune response that eliminates every single infected cell.
Mutations that confer resistance to CTLs are prevalent in the latent reservoir147, 148 and present a formidable but not insurmountable challenge that might be overcome in the setting of a small reservoir in which the restriction of viral escape is enforced by ART, by a vaccine that targets carefully chosen conserved, autologous or polyclonal epitopes, or by novel strategies.
Several therapeutic HIV-1 vaccines have been tested, including whole inactivated virus, recombinant proteins or viruses, DNA vectors or dendritic cell presentation of autologous antigens (reviewed in Ref. 149) (Fig. 3a). Some vaccines improved HIV-1-specific immune responses150, 151, but none so far has enabled sustained interruption of ART, and this metric may be inappropriately stringent for the goal of eradication in the setting of ongoing ART and anti-latency therapies. Furthermore, such vaccines have not yet been tested for the most relevant factors in the context of eradication strategies, such as: the recognition of relevant epitopes in the context of infection emerging from the latent state; a reduction of low-level viraemia that persists during ART; or a decrease in the frequency of latently infected cells. These aspects should be considered in future studies.
Cell-based therapies. One potential strategy to clear persistently infected cells is the adoptive transfer of HIV-1-specific CTLs152. In a primary cell model of latency, Gag-stimulated CTLs are much more effective at clearing reactivated HIV-1-infected cells than freshly isolated CD8+ T cells144. Preliminary work shows that expansion of T cells against multiple overlapping peptides from different HIV-1 antigens enables increased clearance of reactivated latently infected cells ex vivo153.
Other cytotoxic immune cells are of interest owing to their potential to clear infected cells, in some cases using mechanisms that complement the action of CTLs154. Natural killer cells, lymphokine-activated killer (LAK) cells155 and γδ T cells156 are also very effective at eliminating virally infected targets. However, similarly to CTLs, these immune effectors require priming for optimal function. In oncology applications, effector cells are primed with cytokines, such as IL-15, that have been administered directly in vivo or used ex vivo for activation before reinfusion157, 158. Monoclonal antibodies could also improve effector cell engagement with the infected targets and, in the case of natural killer cells, mediate targeted lysis via antibody-dependent cellular cytotoxicity (ADCC)159, 160 (Fig. 3b).
Gene therapy. Effector cells can also be genetically engineered to increase their efficiency and redirect them to the desired targets. Such approaches have been pioneered in oncology, whereby T cells are genetically modified to express chimeric antigen receptors (CARs) with improved antigen specificity161. This strategy has been adapted to target HIV-1 by genetically modifying peripheral blood cells with a molecularly cloned TCR that redirects cells to viral antigens (Fig. 3c). Encouraging results were shown in a study in which a TCR from a patient who had a sustained and robust CTL response against the HIV-1 p17 Gag-derived antigen SL9 was cloned and expressed in primary CD8+ cells162, and a Phase 1 clinical trial is being carried out (clinicaltrials.gov identifier NCT00991224). Nonetheless, this novel and promising tool should be carefully explored owing to potential off-target toxicities163. Although such approaches might be too resource-intensive to be implemented on a global scale, they may provide proof-of-concept that could lead to strategies that are appropriate for global implementation.
Reversing immune exhaustion. Chronic HIV-1 infection leads to the upregulation of inhibitory co-receptors, such as PD-1, on T cells164 and cytotoxic T lymphocyte-associated protein 4 (CTLA4) (Ref. 165), which are cellular markers of immune exhaustion91 that have an important role in the ineffective viral immune response. Blockade of the PD-1 pathway reverses this state of exhaustion and restores the ability of T cells to inhibit HIV-1 replication in vitro and in vivo in animal models166 and thus presents a promising potential therapeutic intervention (Fig. 3a) that will soon be tested in a clinical trial (clinicaltrials.gov identifier NCT02028403).
Immunotoxins and radioimmunotherapy. Immunotoxins are bifunctional chimeric proteins that consist of a targeting portion, such as an antibody or a ligand, and a toxin effector domain167. Initial clinical trials using immunotoxins did not have a sustained impact on immunological or clinical endpoints168, perhaps owing to the lack of support from an ART regimen. The addition of the immunotoxin 3B3-PE38 (Ref. 169) to ongoing ART was recently shown to reduce tissue levels of HIV-1 RNA in a humanized mouse model102 to several logs below the levels that are seen with ART alone (Fig. 3d). Moreover, radioimmunotherapy using radiolabelled antibodies that target the HIV-1 envelope proteins gp120 and gp41 led to a depletion of chronically HIV-1-infected cells in a severe combined immunodeficiency-peripheral blood lymphocyte (SCID-PBL) mouse model that does not allow viraemia and viral replication170. However such short-term animal studies cannot yet address potential off-target effects, such as hepatotoxicity, that are seen with older immunotoxins168.
Conclusions
Efforts to develop therapies that could eradicate HIV-1 infection or achieve a durable remission of viraemia in the absence of ART have recently accelerated and expanded. Although this initial period of renewed effort has been marked by much progress and enthusiasm, both the scientific and the patient community must be prepared for the prolonged effort that will be required to overcome both the expected and the unforeseen challenges ahead. First, and perhaps most daunting, is the need to target latency within specific cellular reservoirs to disrupt viral quiescence so that residual infection can be cleared (Fig. 4). An alternative strategy would be to permanently repress HIV-1 gene expression or to directly destroy the genome. Recent advances with gene-modifying technologies such as zinc-finger nucleases, TALENs (transcription activator-like effector nucleases) and the CRISPR-Cas (clustered, regularly interspaced short palindromic repeats-CRISPR-associated proteins) system are exciting171, 172. Although these approaches would be more elegant, their implementation would require tremendous advances in gene delivery, as efficient and effective delivery systems to destroy the viral genomes in rare cells throughout the body are not currently available.
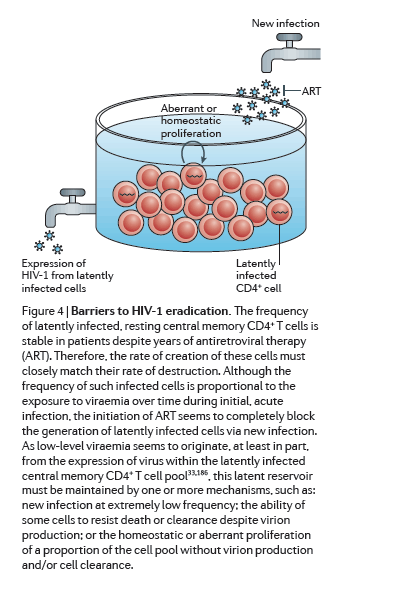
The several approaches to disrupt HIV-1 latency by inducing proviral expression seem to be promising and might be implemented in the foreseeable future. As reactivation of the latent reservoir may be governed by stochastic mechanisms (that is, some latent genomes remain silent even in the event of a single round of maximal mitogen stimulation)138, combinatorial latency-reversing therapy that is safe enough to enable multiple administrations may be needed. Ongoing work aims at designing an effective dosing regimen. Potency must be balanced with minimizing toxicity, and the potential impact of any latency-reversing agent on the immune system must be carefully considered.
In addition, a parallel effort must be made to develop immune-based therapies to ensure and accelerate the process of viral clearance. This effort should be linked to studies of the disruption of latency, as the modalities are likely to be combined. Therefore, given the broad efforts to discover reagents that disrupt latency in resting TCM cells as a first step towards viral eradication therapies, systems must be developed to study the interactions between anti-latency approaches that might be used in combination, as well as the interactions between such agents and immunotherapeutic agents that might be used as a potential cure.
The development of approaches to eradicate HIV-1 infection will take time and durable investment in research towards this goal. It is possible that therapies that result in the depletion of persistent infection and the augmentation of the immune response might lead to an intermediate result. Termed by many as a 'functional cure', this is a state in which HIV-1 infection is not cleared but is so tightly controlled by the immune response that the patient is no longer infectious and is clinically stable in the absence of ART. If so, to make such an investment superior to once-a-day ART (or less with the long-acting therapeutic agents that are currently under development), interventions that result in a functional cure would also have to spare patients the chronic immune activation that is seen in natural 'elite controllers' of HIV-1 infection173, with its attendant risks of long-term morbidity, as otherwise life-long ART might then be clinically preferable.
The scientific and medical challenges in the effort to eradicate HIV-1 infection are formidable and complex. Ultimately, given the scope of the HIV-1 pandemic, strategies to eradicate the disease must be implemented globally. However, to move the field forwards, early proof-of-concept studies are likely to involve approaches that are not widely feasible; for example, bone marrow transplantation or extremely early HIV-1 treatment may never be practical, but the successes and failures of these approaches can provide valuable insights. Although disappointing, the very recent viral rebound that was observed in the Mississippi baby1 after more than 2 years after ART interruption provides valuable clues. Viral rebound after such a long period of time without viraemia in the absence of measurable HIV-1-specific immunity suggests, akin to the Boston patients2, that individual latently infected cells may remain virologically dormant for a considerable period of time before generating viraemia. The short duration of aviraemia in the Boston patients compared with the longer time off therapy in the Mississippi baby case might simply reflect a lower number of latently infected cells or some immune protection conferred during initial exposure to HIV-1 in the Mississippi baby. Approaches to disrupt latency, or even robustly enforce latency, may succeed if the infected cell population is small enough and durable mechanisms to enhance the HIV-1 immune response are present.
As efforts advance, additional obstacles to clear HIV-1 infection are likely to be uncovered, and careful consideration must be given to the ethics of translational research with otherwise healthy volunteers infected with HIV-1. The recent reinvigoration of efforts to gain a detailed understanding of the biology and pathogenesis of viral latency should give hope that we can overcome these obstacles. The journey towards a cure for HIV/AIDS has begun.
|
|
| |
| |
|
|
|