| |
Ombitasvir plus paritaprevir plus ritonavir with or without ribavirin in treatment-naive and treatment-experienced patients with genotype 4 chronic hepatitis C virus infection (PEARL-I): a randomised, open-label trial
|
| |
| |
Download the PDF her
86 patients were treatment-naive, of whom 44 received ombitasvir plus paritaprevir plus ritonavir and 42 received ombitasvir plus paritaprevir plus ritonavir with ribavirin, and 49 treatment-experienced patients received the ribavirin-containing regimen (figure 1). 68 (50%) of 135 patients were infected with viral subtype 4d and 50 (37%) with subtype 4a based on phylogenetic analysis. Demographic characteristics were similar across treatment groups (table 1). 23 (47%) of 49 treatment-experienced patients were null responders.
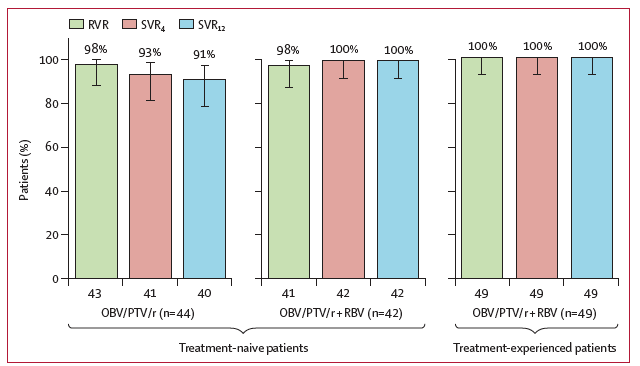
In treatment-naive patients, SVR12 rates were 100% (42/42 [95% CI 91·6-100]) in the ribavirin-containing regimen and 90·9% (40/44 [95% CI 78·3-97·5]) in the ribavirin-free regimen (figure 2); there was no statistical difference in SVR12 rates between these two treatment groups after adjusting for interleukin 28B genotype (mean difference -9·16% [95% CI -19·61 to 1·29]; p=0·086). All treatment-experienced patients (49/49; 100% [95% CI 92·7-100]) in the ribavirin-containing group achieved SVR12.
No relapses between post treatment week 12 and post treatment week 24 have been recorded in treatment-naive patients in either treatment group; the treatment-experienced patients have not yet reached post treatment week 24, but no relapses have been observed after post treatment week 12 in this group of patients.
Three treatment-naive patients who received ombitasvir plus paritaprevir plus ritonavir without ribavirin had virological failure: one patient (2%) of 44 had virological breakthrough at treatment week 8, and two (5%) of 42 relapsed before post treatment week 12. Adherence data were not available for the patient who had virological breakthrough at treatment week 8; adherence was high (>90%) for the two patients who had post treatment relapse (assessed via medication event monitoring systems). All three patients were infected with subtype 4d, and all had resistance-associated variants present at the time of failure that were not present at baseline. The predominant variants in NS3 and NS5A were D168V and L28S or L28V, respectively. Two of the three patients who had virological failure had CT interleukin 28B, which was the most common genotype observed in the study population; one patient had TT interleukin 28B genotype.
The most common treatment-emergent adverse events were headache (14 [29%] of 49 treatment-experienced patients vs 14 [33%] of 42 treatment-naive patients given combination plus ritonavir), asthenia (10 [24%] vs 16 [33%]), fatigue (3 [7%] vs 9 [18%]), insomnia (2 [5%] vs 8 [16%]), and nausea (4 [9%] vs 7 [17%]; table 2). Of the 114 patients that experienced treatment-emergent adverse events, 111 (97%) experienced treatment-emergent adverse events that were mild in severity. One treatment-naive patient (2%) of 44 who received the ribavirin-free regimen had a serious treatment-emergent adverse event (contusion due to traffic accident) that was considered unrelated to study medication. No patients had treatment-emergent adverse event-related discontinuations or dose interruptions. No laboratory abnormalities above grade 3 were reported.
Overall, three (2%) of 135 patients (one in each treatment group) had haemoglobin concentrations of 80 to less than 100 g/L. One treatment-naive patient assigned to the ribavirin-containing treatment group had a grade 3 haemoglobin value of 65 g/L on day 24 of the study. This patient's haemoglobin value was normal at the next study visit (day 29); no associated adverse events were noted and ribavirin dose was not adjusted. Six (7%) of 91 patients had adverse events leading to ribavirin dose modification. One (2%) of 42 treatment-naive patients and three (6%) of 49 treatment-experienced patients had ribavirin dose reduction for haemoglobin level less than 100 g/dL (anaemia), but none required blood transfusion or erythropoietin. Two treatment-naive patients required ribavirin dose modification for adverse events unrelated to anaemia (one [2%] of 42 for anxiety, palpitations, and insomnia and one [2%] for erythema).
---------------
Ombitasvir plus paritaprevir plus ritonavir with or without ribavirin in treatment-naive and treatment-experienced patients with genotype 4 chronic hepatitis C virus infection (PEARL-I): a randomised, open-label trial
The Lancet June 20 2015
Christophe Hezode, Tarik Asselah, K Rajender Reddy, Tarek Hassanein, Marina Berenguer, Katarzyna Fleischer-Stepniewska, Patrick Marcellin,
Coleen Hall, Gretja Schnell, Tami Pilot-Matias, Niloufar Mobashery, Rebecca Redman, Regis A Vilchez, Stanislas Pol
Summary
Background
Hepatitis C virus (HCV) genotype 4 accounts for about 13% of global HCV infections. Because interferon-containing treatments for genotype 4 infection have low efficacy and poor tolerability, an unmet need exists for effective all-oral regimens. We examined the efficacy and safety of an all-oral interferon-free regimen of ombitasvir, an NS5A inhibitor, and paritaprevir (ABT-450), an NS3/4A protease inhibitor dosed with ritonavir (ombitasvir plus paritaprevir plus ritonavir), given with or without ribavirin.
Methods
In this multicentre ongoing phase 2b, randomised, open-label combination trial (PEARL-I), patients were recruited from academic, public, and private hospitals and clinics in France, Hungary, Italy, Poland, Romania, Spain, Turkey, and the USA. Eligible participants were aged 18-70 years with non-cirrhotic, chronic HCV genotype 4 infection (documented ≥6 months before screening) and plasma HCV RNA levels higher than 10 000 IU/mL. Previously untreated (treatment-naive) patients were randomly assigned (1:1) by computer-generated randomisation lists to receive once-daily ombitasvir (25 mg) plus paritaprevir (150 mg) plus ritonavir (100 mg) with or without weight-based ribavirin for 12 weeks. Previously treated (treatment-experienced) patients who had received pegylated interferon plus ribavirin all received the ribavirin-containing regimen. The primary endpoint was a sustained virological response (HCV RNA <25 IU/mL) 12 weeks after the end of treatment (SVR12). Analysis was by intention to treat. This study is registered with ClinicalTrials.gov, number NCT01685203.
Findings
Between Aug 14, 2012, and Nov 19, 2013, 467 patients with HCV infection were screened, of whom 174 were infected with genotype 4. 135 patients were randomly assigned to treatment and received at least one dose of study medication; 86 patients were treatment-naive, of whom 44 received ombitasvir plus paritaprevir plus ritonavir and 42 received ombitasvir plus paritaprevir plus ritonavir with ribavirin, and 49 treatment-experienced patients received the ribavirin-containing regimen. In previously untreated patients, SVR12 rates were 100% (42/42 [95% CI 91·6-100]) in the ribavirin-containing regimen and 90·9% (40/44 [95% CI 78·3-97·5]) in the ribavirin-free regimen. No statistically significant differences in SVR12 rates were noted between the treatment-naive groups (mean difference -9·16% [95% CI -19·61 to 1·29]; p=0·086). All treatment-experienced patients achieved SVR12 (49/49; 100% [95% CI 92·7-100]). In the ribavirin-free group, two (5%) of 42 treatment-naive patients had virological relapse, and one (2%) of 44 had virological breakthrough; no virological failures were recorded in the ribavirin-containing regimen. The most common adverse event was headache (14 [29%] of 49 treatment-experienced patients and 14 [33%] of 42 treatment-naive patients). No adverse event-related discontinuations or dose interruptions of study medications, including ribavirin, were noted, and only four patients (4%) of 91 receiving ribavirin required dose modification for haemoglobin less than 100 g/L or anaemia.
Interpretation
An interferon-free regimen of ombitasvir plus paritaprevir plus ritonavir with or without ribavirin achieved high sustained virological response rates at 12 weeks after the end of treatment and was generally well tolerated, with low rates of anaemia and treatment discontinuation in non-cirrhotic previously untreated and previously treated patients with HCV genotype 4 infection.
Funding
AbbVie.
Introduction
Chronic hepatitis C virus (HCV) infection is a global health problem, with 130-150 million people infected worldwide.1 The infection is a common cause of chronic progressive liver disease (eg, cirrhosis)2 and hepatocellular carcinoma.3 At least seven HCV genotypes (genotypes 1-7) and 67 subtypes have been identified;4 their prevalence rates differ by geographical region. Globally, HCV genotype 4 accounts for roughly 13% of all HCV infections.5 HCV genotype 4 is common in the Middle East, north Africa, and sub-Saharan Africa and is responsible for more than 90% of HCV infections in Egypt.5 In Europe, prevalence of HCV genotype 4 accounts for 14-20% of HCV infections in some countries.5 However, the worldwide prevalence of genotype 4 is based on serological data and might not reflect the actual number of patients infected with the virus.
New direct-acting antiviral drugs, such as the protease inhibitor simeprevir and the nucleotide polymerase inhibitor sofosbuvir, have been suggested for use in genotype 4-infected patients.6 However, guidelines advise the use of these new therapies in combination with pegylated interferon, ribavirin, or both. Such regimens might be poorly tolerated because of symptoms associated with pegylated interferon (flu-like symptoms, psychiatric symptoms, and fatigue) and ribavirin (haematological side-effects, such as anaemia).7, 8 Consequently, a clear unmet need exists for potent, all-oral direct-acting antiviral regimens that can increase the likelihood of treatment success with a more favourable tolerability profile.
The introduction of all-oral, interferon-free regimens that combine direct-acting antiviral drugs has significantly advanced the treatment of HCV, especially for patients with HCV genotype 1 infection.9 High efficacy rates (greater than 95%), low rates of treatment discontinuation, and favourable adverse event profiles have been shown with multiple regimens, both with and without ribavirin.10, 11, 12, 13, 14 However, efficacy and safety data of direct-acting antiviral drugs in patients with HCV genotype 4 infection are scarce.15
Ombitasvir (formerly ABT-267), a potent NS5A inhibitor, and paritaprevir (formerly ABT-450), a potent NS3/4A protease inhibitor identified for clinical development by AbbVie and Enanta, both show in-vitro antiviral activity against HCV genotypes 1a, 1b, 2a, 3a, 4a, and 6a.16, 17, 18 Paritaprevir is given with low-dose ritonavir to increase paritaprevir peak and trough concentrations and overall drug exposure.19 In phase 3 trials, combination therapy with ombitasvir plus paritaprevir plus ritonavir, and dasabuvir (a non-nucleoside NS5B polymerase inhibitor), with and without ribavirin, showed efficacy and safety in treatment-naive and treatment-experienced HCV genotype 1-infected patients with or without compensated cirrhosis.14, 20, 21
We aimed to assess the safety and efficacy of an all-oral, interferon-free regimen of ombitasvir plus paritaprevir plus ritonavir with and without ribavirin in HCV genotype 1b-infected treatment-naive and pegylated interferon plus ribavirin treatment-experienced patients with and without cirrhosis and genotype 4-infected treatment-naive and pegylated interferon plus ribavirin treatment-experienced patients without cirrhosis. The rationale for examining this combination regimen in HCV genotype 4-infected patients was based on the comparable in-vitro potency of these two direct-acting antiviral drugs for HCV genotypes 1b and 4a.16, 17, 18Although dasabuvir was administered as part of the regimen studied in phase 3 clinical trials in genotype 1-infected patients, it was not included in this study because this drug has no activity against genotype 4.
Methods
Study design and participants
In this multicentre ongoing phase 2b, randomised, open-label combination study (PEARL-I), patients were recruited from academic, public, and private hospitals and clinics in France, Hungary, Italy, Poland, Romania, Spain, Turkey, and the USA. The study was designed as an open-label study to maximise the probability of all genotype 4-infected patients in the study achieving sustained virological response. Additionally, an active comparator group that contained pegylated interferon was not included because it could not be effectively blinded. All genotype 1b-infected patients without cirrhosis were enrolled and completed treatment before enrolment of the genotype 4-infected treatment-naive patients to allow for a sequential evaluation of the two-direct-acting antiviral drug regimen in these two patient populations. The first participant was screened in August, 2012, and the last participant completed treatment in March, 2014. All ongoing patients are in post treatment follow-up. The results reported here are from the primary database lock, which was completed when all patients had reached post treatment week 12.
Patients enrolled were aged 18-70 years with chronic HCV genotype 4 infection (documented ≥6 months before screening) and plasma HCV RNA levels greater than 10 000 IU/mL. Enrolled patients were non-cirrhotic, as shown by a liver biopsy within 24 months before or during screening, a FibroTest score 0·72 or less and aspartate aminotransferase (AST):platelet index 2 or less, or a screening FibroScan result less than 9·6 kPa. Patients were treatment-naive or had previously received pegylated interferon plus ribavirin therapy and met criteria for a null responder, partial responder, or relapser (appendix). Exclusion criteria included positive results at screening for hepatitis B surface antigen or anti-HIV antibodies, other causes of liver disease, or current or past clinical evidence of cirrhosis.
The study was approved by all institutional review boards and conducted in accordance with the International Conference on Harmonisation guidelines and the Declaration of Helsinki. Written informed consent was obtained from each patient before enrolment.
Randomisation and masking
Genotype 4-infected treatment-naive patients were stratified by host genetic background and interleukin 28B genotype (favourable, CC genotype vs unfavourable, non-CC genotype) and were randomly assigned (1:1) to receive once-daily ombitasvir plus paritaprevir plus ritonavir with or without twice-daily, weight-based ribavirin. Randomised treatments were assigned by the interactive response technology vendor with computer-generated randomisation lists prepared by the funder's randomisation personnel.
Procedures
Based on a protocol-specified interim review of the results from the first ten genotype 4-infected treatment-naive patients who received ombitasvir (25 mg) plus paritaprevir (150 mg) plus ritonavir (100 mg) with or without twice-daily, weight-based ribavirin, which indicated higher efficacy rates for patients who received the ribavirin-containing regimen, genotype 4-infected treatment-experienced patients were enrolled and received once-daily ombitasvir (25 mg) plus paritaprevir (150 mg) plus ritonavir (100 mg) with twice-daily weight-based ribavirin. All patients who received at least one dose of study drug participated in the post treatment phase, during which sustained virological response and the emergence and persistence of resistant viral variants in those who failed therapy were monitored. For assessments of sustained virological response, patients with no HCV RNA values (eg, patients who were lost to follow-up) were counted as failures.
Extraction of HCV RNA from plasma samples and quantification of HCV RNA levels were done by a central laboratory. Extraction was done with the Roche High Pure System Viral Nucleic Acid Kit (Roche, Nutley, NJ, USA); HCV RNA levels were determined with the COBAS TaqMan real-time reverse-transcriptase polymerase chain reaction assay 2·0 (Roche), which has a lower limit of quantitation of 25 IU/mL.
The Versant HCV Genotype Inno-LiPA assay (LiPA) was used to determine HCV genotype for patients in this study; however, the LiPA assay was unable to accurately identify the subtype for HCV genotype 4,22 because it is a heterogeneous genotype with roughly 17 subtypes.4 Therefore, the viral subtype for HCV genotype 4-infected patients was determined by phylogenetic analyses of NS5B (a 329 nucleotide region)23 and full-length NS3/4A and NS5A nucleotide sequences. For patients who did not achieve sustained virological response (HCV RNA <25 IU/mL 12 weeks after the last dose of study drug [SVR12]), sequences of HCV NS3/4A and NS5A at baseline and at the time of failure were determined by population nucleotide sequencing, and translated NS3/4 and NS5A aminoacid sequences were used to identify treatment-emergent variants.
Adverse events were assessed at every study visit from the time of first study drug administration until 30 days post treatment. Serious adverse events were monitored throughout the study period. All adverse events were coded according to the Medical Dictionary for Regulatory Activities version 12.1. The severity and relation of adverse events to study drug in the opinion of the investigator were reported. Clinical and laboratory parameters were assessed throughout the study.
Outcomes
The primary protocol-specified efficacy endpoint was SVR12. Secondary protocol-specified efficacy endpoints included post treatment relapse, on-treatment virological failure, rate of sustained virological response 4 weeks after the last dose of study drug (SVR4), and rapid virological response (appendix).
Statistical analysis
Based on in-vitro data, which indicated similar antiviral activity for ombitasvir and paritaprevir for genotype 1b and 4a,16, 17, 18 it was expected that in this study, sustained virological response rates in patients with genotype 4 infection would be similar to those of genotype 1b-infected patients; thus, the sample size calculations for the genotype 1b-infected and genotype 4-infected patients without cirrhosis assumed that 70% of treatment-experienced and 95% of treatment-naive patients without cirrhosis would achieve SVR12. This assumption indicated that 40 patients per group would provide roughly 80% power with Fisher's exact test with a two-sided significance level of 0·05 to detect a 25% difference between treatment-naive and treatment-experienced patients.
All randomised patients received at least one dose of study medication and were included in all efficacy and safety analyses (modified intent-to-treat population). The number and percentage of patients achieving each efficacy endpoint were summarised, along with 95% CIs. In the case of a missing HCV RNA value in a defined visit window, the closest values before and after the window were used for flanking imputation. The difference (and corresponding 95% CIs) in SVR12 response between treatment-naive patients receiving the two-direct-acting antiviral drug regimen and those receiving the two-direct-acting antiviral regimen plus ribavirin was estimated with stratum-adjusted Mantel-Haenszel proportions and continuity-corrected variances adjusting for interleukin 28B genotype (CC or non-CC). The number and percentage of patients with treatment-emergent adverse events or potentially clinically significant laboratory or vital sign values were compared between treatment-naive patients who did or did not receive ribavirin with Fisher's exact test.
This study is registered with ClinicalTrials.gov, number NCT01685203.
Role of the funding source
AbbVie funded the study and contributed to study design and conduct; data management, analysis, and interpretation; and the preparation and approval of this report. All authors had access to the study data, reviewed and approved the final report, and take full responsibility for the veracity of the data and statistical analysis. The corresponding author had full access to all study data and had final responsibility for the decision to submit for publication.
Results
Between Aug 14, 2012, and Nov 19, 2013, 467 patients with HCV infection were screened, of whom 174 were infected with genotype 4. 135 patients were enrolled and received at least one dose of study medication; 86 patients were treatment-naive, of whom 44 received ombitasvir plus paritaprevir plus ritonavir and 42 received ombitasvir plus paritaprevir plus ritonavir with ribavirin, and 49 treatment-experienced patients received the ribavirin-containing regimen (figure 1). 68 (50%) of 135 patients were infected with viral subtype 4d and 50 (37%) with subtype 4a based on phylogenetic analysis. Demographic characteristics were similar across treatment groups (table 1). 23 (47%) of 49 treatment-experienced patients were null responders. Two premature discontinuations occurred in treatment-naive patients who received the ribavirin-free regimen; these patients were counted as treatment failures (figure 1).
In treatment-naive patients, SVR12 rates were 100% (42/42 [95% CI 91·6-100]) in the ribavirin-containing regimen and 90·9% (40/44 [95% CI 78·3-97·5]) in the ribavirin-free regimen (figure 2); there was no statistical difference in SVR12 rates between these two treatment groups after adjusting for interleukin 28B genotype (mean difference -9·16% [95% CI -19·61 to 1·29]; p=0·086). All treatment-experienced patients (49/49; 100% [95% CI 92·7-100]) in the ribavirin-containing group achieved SVR12. Rates of rapid virological response and SVR4 were similar or numerically higher in treatment-naive patients who received the ribavirin-containing regimen compared with those who did not receive ribavirin (figure 2). No relapses between post treatment week 12 and post treatment week 24 have been recorded in treatment-naive patients in either treatment group; the treatment-experienced patients have not yet reached post treatment week 24, but no relapses have been observed after post treatment week 12 in this group of patients.
Three treatment-naive patients who received ombitasvir plus paritaprevir plus ritonavir without ribavirin had virological failure: one patient (2%) of 44 had virological breakthrough at treatment week 8, and two (5%) of 42 relapsed before post treatment week 12. Adherence data were not available for the patient who had virological breakthrough at treatment week 8; adherence was high (>90%) for the two patients who had post treatment relapse (assessed via medication event monitoring systems). All three patients were infected with subtype 4d, and all had resistance-associated variants present at the time of failure that were not present at baseline. The predominant variants in NS3 and NS5A were D168V and L28S or L28V, respectively. Two of the three patients who had virological failure had CT interleukin 28B, which was the most common genotype observed in the study population; one patient had TT interleukin 28B genotype.
The most common treatment-emergent adverse events were headache (14 [29%] of 49 treatment-experienced patients vs 14 [33%] of 42 treatment-naive patients given combination plus ritonavir), asthenia (10 [24%] vs 16 [33%]), fatigue (3 [7%] vs 9 [18%]), insomnia (2 [5%] vs 8 [16%]), and nausea (4 [9%] vs 7 [17%]; table 2).
Of the 114 patients that experienced treatment-emergent adverse events, 111 (97%) experienced treatment-emergent adverse events that were mild in severity. One treatment-naive patient (2%) of 44 who received the ribavirin-free regimen had a serious treatment-emergent adverse event (contusion due to traffic accident) that was considered unrelated to study medication. No patients had treatment-emergent adverse event-related discontinuations or dose interruptions. No laboratory abnormalities above grade 3 were reported.
Overall, three (2%) of 135 patients (one in each treatment group) had haemoglobin concentrations of 80 to less than 100 g/L. One treatment-naive patient assigned to the ribavirin-containing treatment group had a grade 3 haemoglobin value of 65 g/L on day 24 of the study. This patient's haemoglobin value was normal at the next study visit (day 29); no associated adverse events were noted and ribavirin dose was not adjusted. Six (7%) of 91 patients had adverse events leading to ribavirin dose modification. One (2%) of 42 treatment-naive patients and three (6%) of 49 treatment-experienced patients had ribavirin dose reduction for haemoglobin level less than 100 g/dL (anaemia), but none required blood transfusion or erythropoietin. Two treatment-naive patients required ribavirin dose modification for adverse events unrelated to anaemia (one [2%] of 42 for anxiety, palpitations, and insomnia and one [2%] for erythema).
In all treatment groups, alanine aminotransferase (ALT) and AST concentrations improved from baseline beginning at week 1 and continued through the last protocol-specified laboratory assessment at post treatment week 4 (appendix).
One patient had an asymptomatic AST elevation at one visit (table 3), which resolved spontaneously with continued dosing. No concomitant elevations greater than grade 2 were noted for ALT levels or bilirubin concentrations. Three treatment-experienced patients had grade 3 bilirubin concentrations; ALT and international normalised ratio of these patients were normal. Three patients with hyperbilirubinaemia had a bilirubin elevation at a single visit, which decreased or normalised with continued treatment. These bilirubin elevations were mainly indirect and probably related to the combined effects of ribavirin-associated hemolysis and paritaprevir on the bilirubin transporter OATP-1. No concomitant ALT or AST elevations were observed.
Discussion
HCV genotype 4 infections account for a large proportion of the worldwide HCV epidemic.24 In this study of an all-oral, interferon-free, 12-week regimen of ombitasvir plus paritaprevir plus ritonavir with or without ribavirin, high SVR12 rates were achieved in HCV genotype 4-infected treatment-naive and treatment-experienced patients without cirrhosis. Although no difference between the ribavirin-containing and ribavirin-free regimens was noted, the 100% (42/42) SVR12 rate recorded with the ribavirin-containing regimen in treatment-naive and treatment-experienced patients suggests that this multitargeted regimen provides the highest certainty of achieving sustained virological response in patients infected with diverse HCV genotype 4 subtypes (panel). The addition of ribavirin to the two-direct-acting antiviral drug regimen could be an important consideration for physicians when treating patients with HCV genotype 4 because subtyping is not a common clinical procedure, and genotype 4 is a heterogeneous genotype with multiple subtypes.4 Unfortunately, we do not know the susceptibility of all subtypes to direct-acting antiviral drugs, and larger clinical studies are necessary to fully understand the role of ribavirin in this regimen.
Panel
Research in context
Systematic review
We searched PubMed and meeting abstracts from the European Association for the Study of the Liver and the American Association for the Study of Liver Diseases up to Nov 14, 2014, for clinical studies including patients with chronic hepatitis C virus (HCV) genotype 4 infection, using the search terms "hepatitis C virus" and "HCV" and "genotype". At present, recommended treatment for patients with genotype 4 infection includes the direct-acting antiviral drugs simeprevir or sofosbuvir in combination with pegylated interferon with or without ribavirin for all interferon-eligible patients.6 Two ongoing studies are testing the efficacy and safety of all-oral, interferon-free direct-acting antiviral drug regimens with and without ribavirin in patients with HCV genotype 4 to determine whether high viral response rates can be achieved without the toxicities associated with interferon-based therapy. Only preliminary evidence is available from these studies, both of which include treatment-naive and treatment-experienced patients, some of whom are cirrhotic or have advanced fibrosis. One study is investigating sofosbuvir with ribavirin in Egyptian patients; sustained virological response rates were 77% at 12 weeks and 90% at 24 weeks.29 A second ongoing study is examining sofosbuvir and ledipasvir; of four patients assessed, 75% achieved sustained virological response with 12 weeks of treatment.30
Interpretation
The results of our study suggest that this multitargeted direct-acting antiviral drug regimen, with or without ribavirin, can achieve high rates of sustained virological response in patients with HCV genotype 4 regardless of subtype and is generally well tolerated, with low rates of anaemia and treatment discontinuation. Whether ribavirin needs to be added to the ombitasvir plus paritaprevir plus ritonavir treatment regimen remains an open question. Although treatment-naive patients randomly assigned to receive ombitasvir plus paritaprevir plus ritonavir with ribavirin achieved higher sustained virological response rates than those who received the two-direct-acting antiviral drug regimen without ribavirin, the difference was not significant.
In the past, combination pegylated interferon plus ribavirin was recommended for treatment of HCV genotype 4-infected patients, but this regimen had major limitations, including suboptimum response rates,25 large side-effects, high treatment discontinuation rates, and prolonged treatment durations. Combination therapy with pegylated interferon plus ribavirin and a direct-acting antiviral drug has increased the efficacy of pegylated interferon plus ribavirin-based regimens in genotype 4-infected patients; however, the adverse effects of these currently recommended regimens suggest that effective pegylated interferon-free regimens with more favourable tolerability would be beneficial. The treatment in this trial showed SVR12 rates greater than or similar to those reported with combinations of pegylated interferon plus ribavirin and a direct-acting antiviral drug.26, 27, 28 Other interferon-free direct-acting antiviral drug regimens (eg, sofosbuvir plus ledipasvir and sofosbuvir plus ribavirin) are also being assessed in patients with genotype 4 infection;29, 30 however, published data for the efficacy and safety of these regimens are scare. In a study of genotype 4-infected Egyptian patients with and without cirrhosis who received sofosbuvir plus ribavirin, SVR12 rates were 77% (40/52) with 12 weeks of treatment and 90% (46/51) with 24 weeks.29 In a second ongoing study (NIAID SYNERGY) that examined sofosbuvir and ledipasvir in patients with genotype 4 infection (some of whom had advanced fibrosis), SVR12 rates were 75% (three of four patients) after 12 weeks of treatment.30
In the previous era of interferon-containing therapies, the most powerful pretreatment predictor of sustained virological response in patients with genotype 1 and genotype 4 infection was the favourable CC interleukin 28B genotype.31 In the era of all-oral direct-acting antiviral drug therapies, the positive effect of the CC interleukin 28B genotype varies by treatment regimen.21, 32 However, the effect of the interleukin 28B genotype on sustained virological response rates among genotype 4-infected patients is currently unknown. In this study, such rates were high in most patients that possessed a non-CC interleukin 28B genotype (ie, 90·9-100%), suggesting that this host genotype does not have a restricted effect on response with the regimen of ombitasvir plus paritaprevir plus ritonavir.
Overall, ombitasvir plus paritaprevir plus ritonavir with or without ribavirin was well tolerated, with no study drug discontinuations or interruptions due to treatment-emergent adverse events or treatment-related serious adverse events. Although HCV treatment regimens that contain pegylated interferon plus ribavirin have been associated with substantial haematological abnormalities that lead to treatment interruption or discontinuation or the use of adjuvant therapies (eg, erythropoietin or blood transfusion), in this study only four patients required ribavirin dose modification for anaemia or a haemoglobin decrease. Other trials of this combination regimen have also shown low rates of anaemia with ribavirin.11, 12, 14, 21 These findings could be related to a brisk ribavirin-related reticulocytosis in the absence of the bone marrow suppressant effects of interferon.
This study has several strengths. It is the largest study to date to assess a direct-acting antiviral drug-only regimen in patients with HCV genotype 4 infection in both treatment-naive and treatment-experienced patients, and it included a range of genotype 4 subtypes based on phylogenetic analysis. The limitations include the exclusion of patients with more advanced liver disease (who traditionally have lower rates of treatment response with interferon-based regimens21) and lack of examination of a ribavirin-free regimen in treatment-experienced patients.
Future trials of this two-direct-acting antiviral drug regimen should be undertaken to assess its efficacy and safety in genotype 4-infected patients with cirrhosis.
|
|
| |
| |
|
|
|