| |
Therapy With Direct-Acting Antivirals for Genotype 3 Patients:
Interferon's Last Gasp? Commentary
|
| |
| |
Download the PDF here
Download the PDF here
Download the PDF here
AND 2 studies published alongside this commentary [articles below] this month for Harvoni in GT3 by Gane In New Zealand and Sofosbuvir+Peg/Rbv by Graham which was presented at this past EASL
Gastroenterology Nov 2015
RAFAELESTEBAN
MARIABUTI
Liver Unit, Department of Internal Medicine
Hospital Universitari Vall d'Hebron and
Centro de Investigacion Biomedica en Red de
Enfermedades Hepaticas y Digestivas (CIBERehd)
Instituto de Salud Carlos III
Madrid, Spain
Hepatitis C virus (HCV) genotypes (GTs) 2 and 3 account for approximately 40% of infections by this virus worldwide.1 Patients with HCV GT3 have more rapid disease progression and are less responsive to treatment than those with GT2,2 and GT3 infection is considered relatively difficult to cure with the available direct-acting antivirals (DAAs).3, 4
In the United States, 2 interferon-free regimens have been approved for HCV GT3, namely, sofosbuvir plus ribavirin for 24 weeks and daclatasvir plus sofosbuvir for 12 weeks. The main limitation of DAA combinations for GT3 is the low sustained virologic response (SVR) rates in patients with cirrhosis, particularly treatment-experienced ones. Another option is the combination of sofosbuvir plus peginterferon and ribavirin for 12 weeks, but it is less attractive because of the adverse effects and monitoring requirements of peginterferon. In Europe, 3 regimens are recommended for HCV GT3: sofosbuvir plus peginterferon and ribavirin for 12 weeks, sofosbuvir and ribavirin for 24 weeks, or sofosbuvir and daclatasvir for 12 weeks in noncirrhotic patients.4, 5Response rates for all-oral regimens in GT3 patients with cirrhosis are well below the 90% rates seen in patients with other HCV GTs. For this reason, sofosbuvir plus peginterferon and ribavirin for 12 weeks, or sofosbuvir plus daclatasvir with or without ribavirin for 24 weeks are preferred in treatment-experienced cirrhotic patients. However, these options have been recommended based on limited information that must be confirmed in further studies.
In this issue of Gastroenterology, 2 important papers offer new data on GT3 patients: Gane et al5 describe the safety and efficacy of ledipasvir/sofosbuvir regimens with or without ribavirin for 12 weeks in a study including 126 GT 3 or 6 patients, and Foster et al6 report the efficacy of sofosbuvir plus ribavirin with or without peginterferon alfa in a population of GT3 patients. Gane et al performed a phase II open-label, uncontrolled trial, in which GT3 treatment-naïve patients were assigned randomly to receive ledipasvir/sofosbuvir with or without ribavirin for 12 weeks, whereas treatment-experienced patients received ledipasvir/sofosbuvir and ribavirin for 12 weeks. SVR12 rates were 100% and 82% in treatment-naïve and experienced patients, respectively, treated with ledipasvir/sofosbuvir and ribavirin. SVR rates were only 64% in treatment-naïve individuals receiving ledipasvir/sofosbuvir without ribavirin, suggesting that ribavirin cannot be omitted from this regimen without a considerable loss of efficacy. Addition of ribavirin to ledipasvir/sofosbuvir reduced the relapse rate from 8% to 0% in treatment-naïve individuals, whereas relapse was 8% in treatment-experienced patients. Patients who relapsed were predominantly males with cirrhosis. Ledipasvir/sofosbuvir has been approved for treating HCV GT3 by the European Medicines Agency (EMEA), but not the US Food and Drug Administration (FDA), and the European Association for the Study of the Liver guidelines do not currently recommend the use of this combination.4
Based on the results of the All-Oral 12-Week Treatment With Daclatasvir Plus Sofosbuvir in Patients With Hepatitis C Virus Genotype 3 Infection (ALLY-3) trial, another NS5A complex inhibitor, daclatasvir, in combination with sofosbuvir has been approved by both the FDA and EMEA for treating GT3 patients.7In ALLY-3, daclatasvir plus sofosbuvir for 12 weeks resulted in SVR rates of 90% in treatment-naive and 86% in treatment-experienced patients. SVR was high in patients without cirrhosis (97% and 94%, respectively) and substantially lower in those with cirrhosis (58% and 69%, respectively). Almost all patients who did not achieve SVR relapsed. The reason for the higher relapse rate in GT3 cirrhotic patients is uncertain. Cirrhosis may affect the therapy response owing to drug-related factors such as changes in drug delivery and metabolism resulting from major disruptions in hepatic blood flow.8 These changes could lead to suboptimal drug levels in some regions of the liver, favoring viral reservoirs. It has been reported that the hepatic steatosis frequently found in GT3 patients may alter drug metabolism.9 Factors related to the host immune response could also contribute to relapse. In patients with cirrhosis, particularly those with low albumin levels, there is an increase in the risk of infection, possibly via prostaglandin E2 inhibition of macrophages.10 In addition, a recent report in noncirrhotic patients has shown that interferon-free therapy normalizes natural killer cell function.11
There are no head-to-head studies comparing ledipasvir/sofosbuvir versus daclatasvir and sofosbuvir. In vitro findings suggest that the antiviral potency of daclatasvir against HCV GT3 is superior to that of ledipasvir.12 The results obtained by Gane et al5 when ribavirin was added to ledipasvir/sofosbuvir are better than would be expected, enabling a reduction in the duration of therapy to 12 weeks in treatment-naïve patients, including those with cirrhosis. However, the study was performed only in New Zeeland and it contains a small number of cirrhotic patients. To put the value of ledipasvir into perspective, Gane et al's results can be viewed against those obtained with sofosbuvir and ribavirin for 12 weeks in GT3 treatment-naïve (Fission study)13 and experienced (Fusion study) patients.14 With all the limitations of this type of comparison, ledipasvir/sofosbuvir for 12 weeks increased SVR rates by 9% relative to sofosbuvir and ribavirin in naïve patients, and with the addition of ribavirin, by approximately 40% in naïve and experienced patients. However, daclatasvir and sofosbuvir without ribavirin for the same duration has provided even higher SVR rates than ledipasvir/sofosbuvir (90%), except in patients with cirrhosis.7 In the early access program in UK in decompensated GT3 patients,15 SVR12 was 59% (36/61) for ledipasvir/sofosbuvir plus ribavirin for 12 weeks and 70% (80/114) for daclatasvir and sofosbuvir plus ribavirin for 12 weeks. SVR rates were lower in the small number of patients receiving the same combination without ribavirin. Several lessons have been learned in studies with DAAs: (1) GT3 patients with cirrhosis are still difficult to cure, (2) ribavirin is needed to increase SVR rates, and (3) lengthy therapy is required, ≥16 or 24 weeks even when using combinations of sofosbuvir and an NS5A inhibitor.
In the phase III, open-label, multicenter study by Foster et al6 included in this issue of Gastroenterology, 48 GT2 treatment-experienced patients and 544 GT3 treatment-naïve and experienced patients were randomized to receive sofosbuvir and ribavirin for either 16 or 24 weeks, or sofosbuvir plus peginterferon and ribavirin for 12 weeks. In GT2, SVR rates were 87% and 100% for 16 and 24 weeks of sofosbuvir and ribavirin, respectively, and 94% for sofosbuvir, peginterferon, and ribavirin for 12 weeks. In GT3 patients, SVR12 was 71% and 84% in those receiving 16 or 24 weeks of sofosbuvir and ribavirin, respectively, and 93% in those receiving sofosbuvir, peginterferon, and ribavirin for 12 weeks. Among the major patient subgroups, including treatment-naïve and treatment-experienced patients, those with and without cirrhosis, and in subgroups by combined treatment history and cirrhosis status, SVR12 rates were least among patients receiving 16 weeks of sofosbuvir and ribavirin and greatest among patients receiving 12 weeks of sofosbuvir plus peginterferon and ribavirin. Eighty-three patients had virologic relapse, 50 (28%) in the group receiving 16 weeks of sofosbuvir and ribavirin, 24 (13%) of those receiving 24 weeks of sofosbuvir and ribavirin, and 9 (5%) patients receiving 12 weeks of sofosbuvir plus peginterferon and ribavirin. Baseline factors associated with relapse were male sex, cirrhosis, and interleukin (IL)-28B non-CC haplotype in patients receiving 16 weeks of sofosbuvir and ribavirin. This results support the preliminary data of a phase II study,16 suggesting that the combination of sofosbuvir, peginterferon, and ribavirin remains the best option in terms of efficacy and duration in GT3 patients with cirrhosis, yielding SVR rates of >85%. SVR12 rates in various studies using the approved DAA combinations are shown in Figure 1.
Figure 1. Sustained virologic response rates with different therapeutic regimens (A) in patients without cirrhosis (B) in patients
with cirrhosis.
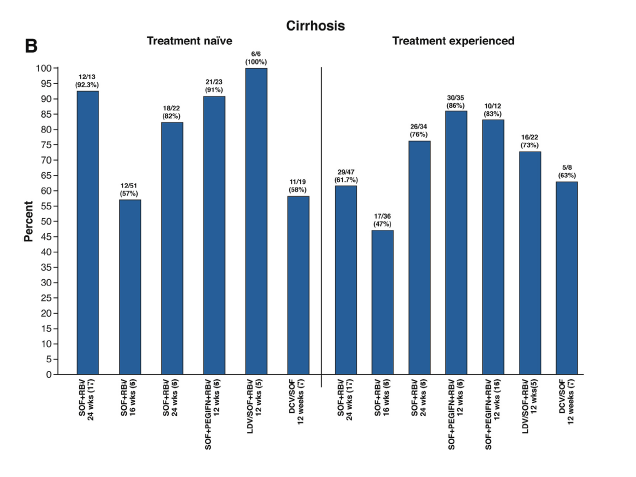
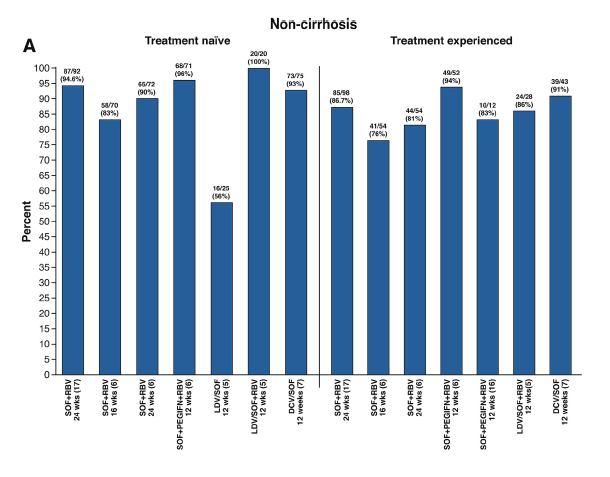
The main drawback of interferon is its associated side effects. Nonetheless, the 12-week regimen of sofosbuvir plus peginterferon and ribavirin in Foster's study unexpectedly showed few differences in terms of adverse events compared with 16 to 24 weeks of sofosbuvir and ribavirin. Some interferon-associated side effects were more common in patients receiving this drug (fatigue, myalgia, flu-like symptoms), but only 1 patient discontinued therapy.6
An advantage of the interferon-based regimen used in this study was that none of the patients who relapsed harbored NS5B resistance-associated variants such as S282T, V321A, or V321A, suggesting that interferon use prevents their emergence. In the sofosbuvir and ribavirin arms, relapsers did not show the S282T resistance-associated variant, but L159F was identified in 8 patients and V321A in 1. Data from Gane et al on RAVs by deep sequencing in the 17 patients who relapsed to ledipasvir/sofosbuvir with or without ribavirin showed no Y93H variants associated with NS5A resistance, whereas the S282T variant was detected in 2 patients.5 This contrasts with previous findings in HCV GT1.17, 18 Both these observations support a lower potency of ledipasvir against HCV GT3. NS5A RAVs (Y93H) were also detected in 9 of 19 patients who failed daclatasvir and sofosbuvir in the ALLY-3 study.7
Emergence of RAVs can compromise salvage therapy, but there is little available information to date on rescue therapy in this scenario. Retreatment with sofosbuvir plus daclatasvir is a potential option in noncirrhotic GT3 patients who fail sofosbuvir and ribavirin, as was seen in 5 of 7 patients included in the ALLY-3 study. However, in treatment-experienced patients (including failure to peginterferon and ribavirin, and sofosbuvir and ribavirin), the best current salvage therapy in those who tolerate interferon is the combination of sofosbuvir, peginterferon, and ribavirin.19 Very few data are available on rescue therapy with sofosbuvir and daclatasvir, but this combination may also have potential efficacy in patients harboring NS5A RAVs.20
Difficult-to-cure HCV GT3 infection may prompt the last revival of interferon-based therapy, in combination with sofosbuvir and ribavirin. For interferon-ineligible patients, ribavirin is an important tool to increase SVR rates in the interim until new, more potent DAAs against GT3 come on the market.
---------------------------------
Efficacy of Ledipasvir and Sofosbuvir, With or Without Ribavirin, for 12 Weeks in Patients With HCV Genotype 3 or 6 Infection
Gastroenterology Nov 2015
Edward J. Gane,1 Robert H. Hyland,2 Di An,2 Evguenia Svarovskaia,2 Phillip S. Pang,2
Diana Brainard,2 and Catherine A. Stedman3
1Auckland Clinical Studies Ltd., Auckland, New Zealand; 2Gilead Sciences, Inc., Foster City, California;
and 3Christchurch Hospital and University of Otago, Christchurch, New Zealand
Background & Aims
We performed a phase 2 clinical trial to evaluate the efficacy and safety of ledipasvir and sofosbuvir, with or without ribavirin, in patients infected with hepatitis C virus (HCV) genotype 3 or 6.
Methods
We performed an open-label study of 126 patients with HCV genotype 3 or 6 infections at 2 centers in New Zealand from April 2013 through October 2014. Subjects were assigned 1 of 4 groups that received 12 weeks of treatment. Previously untreated patients with HCV genotype 3 were randomly assigned to groups given fixed-dose combination tablet of ledipasvir and sofosbuvir (n = 25) or ledipasvir and sofosbuvir along with ribavirin (n = 26).
Treatment-experienced patients with HCV genotype 3 (n = 50) received ledipasvir and sofosbuvir and ribavirin. Treatment-naïve or treatment-experienced patients with HCV genotype 6 (n = 25) received ledipasvir and sofosbuvir. The primary end point was the percentage of patients with HCV RNA ≤15 IU/mL 12 weeks after stopping therapy (sustained virologic response at 12 weeks [SVR12]).
Results
Among treatment-naïve genotype 3 patients, 16 of 25 (64%) receiving ledipasvir and sofosbuvir alone achieved SVR12 compared with all 26 patients (100%) receiving ledipasvir and sofosbuvir and ribavirin. Among treatment-experienced patients with HCV genotype 3, forty-one of fifty achieved an SVR12 (82%). Among patients with HCV genotype 6, the rate of SVR12 was 96% (24 of 25 patients). The most common adverse events were headache, upper respiratory infection, and fatigue. One patient with HCV genotype 3 discontinued ledipasvir and sofosbuvir because of an adverse event (diverticular perforation), which was not considered treatment related.
Conclusions
In an uncontrolled, open-label trial, high rates of SVR12 were achieved by patients with HCV genotype 3 infection who received 12 weeks of ledipasvir and sofosbuvir plus ribavirin, and by patients with HCV genotype 6 infection who received 12 weeks of sofosbuvir and ledipasvir without ribavirin. Current guidelines do not recommend the use of ledipasvir and sofosbuvir, with or without ribavirin, in patients with HCV genotype 3 infection. ClinicalTrials.gov Number: NCT01826981.
Genotype 3 hepatitis C virus (HCV) accounts for approximately 20% of all HCV infections globally and 40% of infections in Asia.1 Although it is frequently grouped with genotype 2 HCV, genotype 3 HCV is associated with greater risk of steatosis,2 fibrosis progression,3 hepatocellular carcinoma,4, 5 and all-cause mortality.4 Genotype 6 HCV constitutes about 1% of HCV infections globally and is found mainly in Southeast Asia and Southern China.6 Genotype 6 HCV is genetically diverse, with 23 subtypes,7 many of which have not been cloned, limiting in vitro testing of antiviral agents. Due to its genetic diversity and relatively low prevalence, genotype 6 HCV is not as well characterized as the other genotypes, but long-term infection appears to be associated with the similar risk of cirrhosis and hepatocellular carcinoma as genotype 1 HCV.8
Sofosbuvir is a potent inhibitor of the HCV NS5B polymerase with a favorable safety profile and a high genetic barrier to resistance.9, 10 In the VALENCE study of treatment-naïve and previously treated patients with and without cirrhosis, sofosbuvir plus ribavirin for 24 weeks resulted in a sustained virologic response at 12 weeks (SVR12) rate of 94% in treatment-naïve patients and 79% in treatment-experienced patients with genotype 3 HCV infection.11 The NEUTRINO study, which evaluated sofosbuvir plus peginterferon-ribavirin in treatment-naïve patients with genotype 1, 4, 5, or 6 HCV, enrolled 6 patients with genotype 6 HCV; all achieved SVR12. The fixed-dose combination of sofosbuvir with ledipasvir, an inhibitor of the HCV NS5A protein, was recently approved for the treatment of treatment-naïve and previously treated patients chronically infected with genotype 1 hepatitis C virus (HCV).12 The efficacy and safety of ledipasvir and sofosbuvir in patients with non-genotype 1 HCV is unknown. In the ION phase 3 trials in which patients with genotype 1 HCV received ledipasvir and sofosbuvir with and without ribavirin for 12 or 24 weeks, patients receiving ledipasvir and sofosbuvir alone had rates of SVR12 similar to those receiving ledipasvir and sofosbuvir plus ribavirin.13, 14, 15 The elimination of ribavirin from the treatment of HCV is desirable due to the adverse events associated with ribavirin therapy.
We evaluated the efficacy and safety of a fixed-dose combination (FDC) tablet of ledipasvir and sofosbuvir administered with or without ribavirin for 12 weeks in treatment-naïve and treatment-experienced patients with genotype 3 or 6 HCV. We included treatment arms with and without ribavirin for treatment-naïve patients with genotype 3 HCV to determine if ribavirin can be omitted from the regimen without a loss of efficacy. At the time the trial was designed, there was no information concerning the efficacy of ledipasvir and sofosbuvir in patients with genotype 3 HCV, so the trial was designed so that the easier-to-treat patients-those naïve to HCV treatment-initiated treatment first, and patients with more difficult to treat genotype 3 HCV-those who had not achieved SVR after previous treatment-would begin ledipasvir and sofosbuvir plus ribavirin only if 90% of the treatment-naïve patients had HCV RNA less than lower limit of quantification (LLOQ) at post-treatment week 4.
Methods
Patients
We enrolled patients at 2 centers in New Zealand during the period from April 2013 to October 2014. Eligible patients were at least 18 years old and had chronic infection with genotype 3 or 6 HCV, with plasma HCV RNA ≥104 IU/mL. The first 2 groups enrolled treatment-naïve patients with genotype 3 HCV, the third group, previously treated patients with genotype 3 HCV, and the fourth group included both treatment-naïve and previously treated patients with genotype 6 HCV. Up to 40% of patients in each group could have compensated cirrhosis, as determined by the following: biopsy, Fibroscan >12.5 kPa, or FibroTest >0.75 and aspartate aminotransferase to platelet ratio index >2. Patients with any of the following characteristics or conditions were excluded from participation: body mass index <18 kg/m2; decompensated liver disease; electrocardiogram with clinically significant abnormalities; chronic use of systemic immunosuppressive or immunomodulatory agents; human immunodeficiency virus infection; hepatitis B virus infection; creatinine clearance <60 mL/min as calculated by the Cockcroft-Gault equation; albumin <3 g/dL; international normalized ratio >1.5 x upper limit of normal unless the patient had known hemophilia or was stable on an anticoagulant regimen affecting international normalized ratio; hemoglobin <11 g/dL for females and <12 g/dL for males; platelets <50,000/mm3; direct bilirubin >1.5 x upper limit of normal, except for patients with Gilbert's syndrome; alanine aminotransferase, aspartate aminotransferase, or alkaline phosphatase >10 x upper limit of normal. All patients provided written informed consent before undertaking any study-related procedures.
Study Design
This was a phase 2, multi-center, open-label study. Patients with HCV genotype 3 were enrolled in 2 parts (Supplementary Figure 1). First, previously untreated patients were randomly assigned in a 1:1 ratio to receive 12 weeks of treatment with ledipasvir and sofosbuvir FDC (90 mg ledipasvir, 400 mg sofosbuvir) once daily or ledipasvir and sofosbuvir FDC once daily plus ribavirin 1000 or 1200 mg/d divided twice daily. The study statistician produced the computer-generated randomization sequence using SAS software (SAS Institute, Cary, NC). Allocation to treatment was done sequentially and Gilead Clinical Operations communicated the randomization sequence to the site by e-mail. If >90% of patients receiving ledipasvir and sofosbuvir plus ribavirin had HCV RNA below the LLOQ at treatment week 4, a third group of HCV genotype 3 patients was to be enrolled. This group of 50 patients who had been previously treated with peginterferon plus ribavirin received 12 weeks of ledipasvir and sofosbuvir FDC once daily plus ribavirin 1000 or 1200 mg/d in a divided dose. A fourth treatment group included treatment-naïve or treatment-experienced patients with HCV genotype 6 who received 12 weeks of ledipasvir and sofosbuvir FDC once daily.
Treatment-experienced patients with HCV genotype 3 and patients with HCV genotype 6 were sequentially assigned to their respective therapies.
The study protocol was approved by each institution's review board or ethics committee before study initiation. The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice Guidelines and the Declaration of Helsinki. The study was designed and conducted by the sponsor in collaboration with the principal investigators. The sponsor collected the data and monitored the study conduct. All authors had access to the study data and reviewed and approved the final article.
Study Assessments
Plasma HCV RNA was analyzed by using the COBAS AmpliPrep/COBAS TaqMan HCV Test, version 2.0 (Roche Molecular Systems, Inc., Branchburg, NJ), which has a LLOQ of 15 IU/mL.
Plasma samples for viral sequencing were collected at the same time points as for HCV RNA levels. Deep sequencing of the HCV NS5A and NS5B-encoding regions was performed for viral samples with HCV RNA ≥1000 IU/mL. Safety data were collected during treatment and for 4 weeks after stopping treatment. The data included reported adverse events, physical examinations, clinical laboratory tests, vital signs, and electrocardiography recordings. Treatment-emergent clinical and laboratory adverse events were summarized using the Medical Dictionary for Regulatory Activities (version 17.0). Virologic relapse was defined as HCV RNA equal to or more than the LLOQ during the post-treatment period in a patient who had HCV RNA less than LLOQ at end of treatment.
End Points and Statistical Analyses
The primary efficacy end point was the percentage of patients in each study group with SVR12, defined as HCV RNA less than LLOQ (15 IU/mL) 12 weeks after stopping study drug. In the primary efficacy analysis, the SVR12 rate was calculated with a 2-sided 95% exact confidence interval (CI) using the Clopper-Pearson method. We calculated that with 25 patients in a treatment group, a 2-sided 95% exact CI would extend at most 42% in length. With 50 subjects in a treatment group, a 2-sided 95% exact CI will extend at most 29% in length. This study was not designed to evaluate formal statistical hypotheses. No inferential statistics or statistical comparisons were planned.
Results
Study Population
In total, 126 patients were enrolled and treated at 2 sites in New Zealand between April 2013 and October 2014. Overall, 71% of patients were white and 63% were male (Table 1). In patients with genotype 3 HCV, the presence of cirrhosis was more common among treatment-experienced (44%) than treatment-naïve (20%) patients. Among the 25 patients with genotype 6 HCV, only 2 had been treated previously and 2 had cirrhosis at baseline. Patients with genotype 6 HCV infection were from the following countries of origin: Cambodia (n = 11), China (n = 7), Myanmar (n = 3), Vietnam (n = 1), and other (n = 3). Figure 1 shows patient disposition throughout the study.
Antiviral Response
Among treatment-naïve genotype 3 patients, the overall rates of SVR12 were 64% (16 of 25) in those receiving ledipasvir and sofosbuvir alone (95% CI: 43%-82%) and 100% (26 of 26) in those receiving ledipasvir and sofosbuvir plus ribavirin (95% CI: 87%-100%), including 6 patients with cirrhosis (Table 2). Two patients with genotype 3 HCV who were receiving ledipasvir and sofosbuvir plus ribavirin had their participation in the study halted after completing at least 8 weeks of treatment due to nonadherence with the visit schedule and dosing requirements; both achieved SVR12. Table 3 shows the baseline characteristics of the 8 patients receiving ledipasvir and sofosbuvir without ribavirin who relapsed.
Of the 50 treatment-experienced patients with genotype 3 HCV receiving 12 weeks of ledipasvir and sofosbuvir plus ribavirin, 41 (82%) achieved SVR12 (95% CI: 69%-91%). The rate of SVR12 was 73% (95% CI: 50%-89%) and 89% (95% CI: 72%-98%) in those with and without cirrhosis, respectively. Of the 9 patients who did not have SVR12, 1 had virologic breakthrough on treatment after having HCV RNA less than LLOQ, and 8 had virologic relapse after treatment. Baseline characteristics of these patients are given in Table 3.
Among the 25 patients with genotype 6 HCV, 24 (96%; 95% CI: 80%-100%) reached SVR12 with ledipasvir and sofosbuvir without ribavirin. The single patient who did not reach SVR12 was a 34-year-old male who withdrew consent during week 8 of treatment and therefore did not receive the full course of treatment. He relapsed by post-treatment week 4.
Viral Sequencing
Sequencing of the NS5B region was successfully carried out on baseline samples from 98 of the 101 genotype 3 patients (3 samples could not be amplified for sequencing). Of these 98 patients, 96 were infected with genotype 3a HCV and 2 with genotype 3k HCV. Baseline sequencing of the NS5B region was successful for all 25 patients infected with genotype 6 HCV. The subtype distribution of patients with genotype 6 HCV was as follows: 8 patients had genotype 6a, 6 had genotype 6e, 3 had genotype 6l, 2 had genotype 6m, 3 had genotype 6p, 2 had genotype 6q, and 1 had genotype 6r. The NS5B variants S282T, V321A, and L159F were not observed at baseline in any patient. Sequencing of the NS5A region was successfully carried out on baseline samples from 100 of the 101 patients with genotype 3 HCV and 23 of the 25 patients with genotype 6 HCV. Common NS5A resistance-associated polymorphisms detected at baseline included 30A/V/S/R/T in genotype 3 and 30R/S/A/L and 93T/S in genotype 6. All patients had at least one common NS5A polymorphism at baseline. In addition, L31M and Y93H were observed in 1 and 8 baseline samples, respectively, of the 100 we were able to sequence. A patient with genotype 3k HCV who had been previously treated with pegylated interferon plus ribavirin and who had the L31M variant at baseline experienced post-treatment relapse. Only 1 of the 8 patients with Y93H at baseline experienced post-treatment relapse. This treatment-naïve patient had genotype 3a HCV and received ledipasvir and sofosbuvir alone.
Deep sequencing of the NS5A and NS5B regions was successfully carried out in samples taken at time of virologic failure from 17 and 18, respectively, of the 19 patients who experienced relapse or breakthrough or had detectable HCV after early discontinuation. The NS5A variant Y93H was not detected in any patient who experienced virologic failure. The NS5B variant S282T, which is associated with resistance to sofosbuvir, was detected at relapse in 2 patients who received ledipasvir and sofosbuvir-1 treatment-naïve patient with genotype 3a HCV at week 2 post treatment (90%) and 1 treatment-naïve patient with genotype 6l at week 12 post treatment (98%). Variants associated with NS5A resistance were not detected in either patient. The variant L159F was detected at time of virologic failure in one treatment-experienced patient with genotype 3a HCV at week 12 of treatment with ledipasvir and sofosbuvir plus ribavirin.
Safety
The adverse events reported in this study were consistent with those reported in prior studies of ledipasvir and sofosbuvir and ribavirin.12, 13, 14, 15, 16 The most common adverse events among all treatment groups were headache and upper respiratory tract infection, followed by fatigue and nausea (Table 4). Six patients experienced serious adverse events. Two of the events, upper abdominal pain and abdominal pain, were considered related to treatment. Only 1 patient, a 38-year-old treatment-naïve male with genotype 3 HCV receiving ledipasvir and sofosbuvir alone, discontinued treatment because of an adverse event. The event, diverticular perforation, was not considered related to treatment. Another patient, a 49-year-old, treatment-experienced woman with genotype 3 HCV, discontinued ribavirin after an event of acute agitation. Seven patients experienced anemia; all were taking ribavirin. Six patients had their dose of ribavirin reduced and one had ribavirin interrupted.
Discussion
In this open-label, phase 2 study, all 26 (100%) treatment-naïve patients with genotype 3 HCV who were randomized to receive 12 weeks of ledipasvir and sofosbuvir plus ribavirin achieved SVR12 as compared with only 16 of 25 (64%) patients who were randomized to receive 12 weeks of ledipasvir and sofosbuvir alone. Although this study was not powered for formal comparisons between these groups, these results suggest that the addition of ribavirin improves the efficacy of ledipasvir and sofosbuvir in treatment-naïve patients with genotype 3 HCV.
Less clear is the benefit ledipasvir adds to the efficacy of sofosbuvir with or without ribavirin. Ledipasvir's lower potency in replicons containing HCV genotype 3a (50% effective concentration [EC50] = 168 nM) as compared with those containing genotype 1a (EC50 = 0.031 nM) or 1b (EC50 = 0.004 nM) suggest that it would have minimal activity against genotype 3 HCV (unpublished data).
In the current trial of 12 weeks of ledipasvir and sofosbuvir plus ribavirin in patients with HCV genotype 3 infection, the SVR12 rate was 100% in treatment-naïve and 82% in treatment-experienced patients. In contrast, in the phase 3 studies of 12 weeks of sofosbuvir plus ribavirin in patients with genotype 3 HCV infection, the SVR12 rate was 56% in treatment-naïve patients and only 30% in treatment-experienced patients.17, 18 While cross-study comparisons are not ideal, the higher SVR rates observed in the current trial support further evaluation of the safety and efficacy of ledipasvir-sofosbuvir plus ribavirin in patients with HCV genotype 3 infection. This would test the hypothesis that the in vitro activity of ledipasvir may not capture in its entirety the efficacy of ledipasvir in patients. At any rate, the reason for the higher rates of SVR in patients who received ledipasvir is not clear and warrants further evaluation.
No Y93H variants associated with NS5A resistance emerged in any genotype 3 or 6 patient with virologic failure, in contrast to previous findings in genotype 1 HCV. The S282T variant was detected in 2 of the 18 patients who experienced virologic failure at time of failure. Both of these observations support the lower potency of ledipasvir against genotype 3 and 6 HCV as compared with genotype 1 HCV.
In vitro studies suggest that the antiviral potency of the HCV NS5A inhibitor daclatasvir against genotype 3 HCV is superior to that of ledipasvir.19 In a recent nonrandomized early access program in patients with genotype 3 HCV and decompensated cirrhosis, the SVR rate among a small number of patients receiving ledipasvir and sofosbuvir was 43% (3 of 7), while the SVR rate among an equally small number receiving daclatasvir and sofosbuvir was 71% (5 of 7).20 When ribavirin was added, the SVR12 rate for patients receiving ledipasvir and sofosbuvir was raised to 59% (36 of 61), but the rate among patients receiving daclatasvir and sofosbuvir plus ribavirin was still significantly higher (70% [80 of 114]; P < .05). Current European Association for the Study of the Liver and American Association for the Study of Liver Diseases guidelines recommend daclatasvir and sofosbuvir with or without ribavirin, but not ledipasvir and sofosbuvir with or without ribavirin in patients with GT 3 infection.21, 22 For patients with genotype 6 HCV infection, the results of this study suggest that ledipasvir and sofosbuvir for 12 weeks may be a safe and effective treatment.
This study was limited by the small size of its treatment arms, the small number of sites (n = 2), and by the lack of a control group. The open-label design is not likely to have had any effect on the assessment of efficacy because the end point of SVR is not subject to bias, but it is conceivable that it biased the reporting of safety events. Another limitation of the study was geographic: all patients were enrolled at 2 sites in New Zealand. In light of these limitations, our results, which are not entirely consistent with previous observations, require further confirmation in a larger clinical trial. An open-label trial of 12 weeks of ledipasvir and sofosbuvir plus ribavirin in approximately 100 treatment-naïve patients with genotype 3 HCV has begun enrollment in Canada (NCT02413593).
In summary, this uncontrolled exploratory study of 12 weeks of treatment with the all-oral regimen of ledipasvir and sofosbuvir with ribavirin resulted in a high rate of SVR among treatment-naïve patients with genotype 3 HCV. Treatment-naïve and treatment-experienced patients with genotype 6 HCV achieved a high rate of SVR with 12 weeks of ledipasvir and sofosbuvir alone.
-------------------------------
Efficacy of Sofosbuvir Plus Ribavirin With or Without Peginterferon-Alfa in Patients With Hepatitis C Virus Genotype 3 Infection and Treatment-Experienced Patients With Cirrhosis and Hepatitis C Virus Genotype 2 Infection
Gastroenterology Nov 2015
Graham R. Foster,1 Stephen Pianko,2 Ashley Brown,3 Daniel Forton,4 Ronald G. Nahass,5
Jacob George,6 Eleanor Barnes,7 Diana M. Brainard,8 Benedetta Massetto,8 Ming Lin,8
Bin Han,8 John G. McHutchison,8 G. Mani Subramanian,8 Curtis Cooper,9 and Kosh Agarwal,10
the BOSON Study Group
1Queen Mary University of London, Barts Health, United Kingdom; 2Monash Health and Monash University, Melbourne,
Victoria, Australia; 3Imperial College Healthcare, National Health Service Trust, London, United Kingdom;
4St George's University of London, London, United Kingdom; 5ID Care, Hillsborough, New Jersey; 6Storr Liver Centre,
Westmead Millennium Institute, University of Sydney and Westmead Hospital, Sydney, New South Wales, Australia;
7Nuffield Department of Medicine, Oxford NHIR BRC and representing STOP-HCV, United Kingdom; 8Gilead Sciences,
Foster City, California; 9The Ottawa Hospital, University of Ottawa, Ottawa, Ontario, Canada; and 10Institute of Liver Studies,
King's College Hospital, London, United Kingdom
Background & Aims
We conducted an open-label, randomized, phase 3 trial to determine the efficacy and safety of sofosbuvir and ribavirin, with and without peginterferon-alfa, in treatment-experienced patients with cirrhosis and hepatitis C virus (HCV) genotype 2 infection and treatment-naïve or treatment-experienced patients with HCV genotype 3 infection.
Methods
The study was conducted at 80 sites in Europe, North America, Australia, and New Zealand Patients were randomly assigned (1:1:1) to groups given sofosbuvir and ribavirin for 16 weeks (n = 196); sofosbuvir and ribavirin for 24 weeks (n = 199); or sofosbuvir, peginterferon-alfa, and ribavirin for 12 weeks (n = 197). The primary end point was the percentage of patients with HCV RNA <15 IU/mL 12 weeks after stopping therapy (sustained virologic response [SVR12]). From October 2013 until April 2014, we enrolled and treated 592 patients-48 with genotype 2 HCV and compensated cirrhosis who had not achieved SVR with previous treatments and 544 with genotype 3 HCV (279 treatment-naïve and 265 previously treated). Overall, 219 patients (37%) had compensated cirrhosis. The last post-treatment week 12 patient visit was in January 2015.
Results
Rates of SVR12 among patients with genotype 2 HCV were 87% and 100%, for those receiving 16 and 24 weeks of sofosbuvir and ribavirin, respectively, and 94% for those receiving sofosbuvir, peginterferon, and ribavirin for 12 weeks. Rates of SVR12 among patients with genotype 3 HCV were 71% and 84% in those receiving 16 and 24 weeks of sofosbuvir and ribavirin, respectively, and 93% in those receiving sofosbuvir, peginterferon, and ribavirin. On-treatment virologic failure occurred in 3 patients with HCV genotype 3a receiving sofosbuvir and ribavirin for 24 weeks. The most common adverse events were fatigue, headache, insomnia, and nausea. Overall, 1% of patients discontinued treatment due to adverse events.
Conclusions
Among patients with genotype 3 HCV infection, including a large proportion of treatment-experienced patients with cirrhosis, the combination of sofosbuvir, peginterferon, and ribavirin for 12 weeks produces high rates of SVR.
Treatment-experienced patients with cirrhosis and genotype 2 HCV infection had high rates of SVR in all groups. EudraCT ID 2013-002641-11.
With the approval of second-wave direct-acting antiviral agents, highly effective interferon-free treatment regimens are now available for the majority of patients chronically infected with the hepatitis C virus (HCV).1, 2 However, a number of questions remain concerning the optimal treatment for certain subgroups of patients infected with genotypes 2 and 3 HCV. Historically, these 2 genotypes, which account for approximately 40% of all HCV infections globally,3 have been grouped together in treatment guidelines. However, it is now recognized that patients with genotype 3 HCV have more rapid disease progression, are less responsive to treatment than patients with genotype 2 HCV, and show variable susceptibility to different direct-acting antiviral agents.4, 5, 6, 7 Although most patients with genotype 2 HCV respond well to the interferon-free combination of sofosbuvir and ribavirin for 12 weeks, treatment-experienced patients with genotype 2 HCV, especially those with cirrhosis, have lower rates of response than treatment-naïve patients.8 Current guidelines recommend the following options for patients with genotypes 2 and 3 HCV: sofosbuvir and ribavirin for 12-24 weeks; 12 weeks of sofosbuvir plus peginterferon and ribavirin; or, more recently, 12 weeks of sofosbuvir plus daclatasvir.1, 2 Response rates for all-oral regimens in patients with genotype 3 HCV and cirrhosis have been well below the 90% rates seen in patients infected with other HCV genotypes.8, 9, 10 Alternative options need to be considered. Phase 2 data suggest that sofosbuvir plus peginterferon and ribavirin may have value in patients with genotype 3 HCV,11 but no adequately powered study to inform clinicians about the comparative safety and efficacy of these regimens has been performed to date.
We therefore conducted a large, phase 3 trial to compare the efficacy and safety of sofosbuvir plus ribavirin with and without peginterferon-alfa in treatment-experienced patients with genotype 2 HCV and cirrhosis and in treatment-naïve and treatment-experienced patients with genotype 3 HCV.
Methods
Study Design and Patients
We conducted this randomized, phase 3, open-label trial at 80 sites in the United Kingdom, Australia, the United States, Canada, and New Zealand. Patients were enrolled between October 18, 2013 and April 1, 2014; the last post-treatment week-12 visit was on January 7, 2015. Eligible patients were at least 18 years old and were chronically infected with HCV, with plasma HCV RNA ≥104 IU/mL. There were no upper limits on age or body mass index. Patients were required to have a platelet count of ≥60,000/mm3 and albumin level ≥3.5 g/dL. We enrolled patients with genotype 2 HCV and compensated cirrhosis who had not achieved sustained virologic response (SVR) after prior HCV treatment for at least 12 weeks with an interferon-based regimen, and patients with genotype 3 HCV with and without compensated cirrhosis who had either never previously received treatment for HCV or who had not achieved SVR after previous HCV treatment. A maximum of 50% of patients with genotype 3 HCV infection with cirrhosis were eligible for inclusion. Previously treated patients who had stopped prior treatment prematurely due to an adverse event were not eligible. Presence of cirrhosis was determined by liver biopsy or by a Fibroscan result of >12.5 kPa. Liver biopsy was required for all patients with genotype 3 HCV except those with contraindications (eg, hemophilia). The study was conducted in collaboration with STOP-HCV, a consortium sponsored by the Medical Research Council, United Kingdom, which assisted in the design of the study.
All patients provided written informed consent before undertaking any study-related procedures. The study protocol was approved by each institution's review board or ethics committee before study initiation. The study was conducted in accordance with the International Conference on Harmonization Good Clinical Practice Guidelines and the Declaration of Helsinki.
Procedures
Randomization was conducted by interactive web and voice-response system (Bracket Global, Wayne, PA). The random allocation sequence was list-based, controlled by a seed, and the list had 4800 slots and a block size of 6. Patients were randomized in a 1:1:1 ratio to 1 of 3 treatment regimens: sofosbuvir and ribavirin for 16 weeks, sofosbuvir and ribavirin for 24 weeks, or sofosbuvir plus peginterferon-alfa and ribavirin for 12 weeks. Enrollment of approximately 600 patients was planned, of which 516 would have genotype 3 HCV infection. Approximately equal numbers of treatment-naïve and treatment-experienced HCV genotype 3 patients were to be enrolled.
Stratification into treatment groups was based on HCV genotype and, for patients with genotype 3 HCV, absence or presence of cirrhosis and history of treatment. No specific target was set for the enrollment of patients with genotype 2 HCV.
Sofosbuvir was administered as an oral tablet of 400 mg once daily. Ribavirin was administered orally twice daily, 1000 or 1200 mg/d in a divided dose based on body weight of <75 kg or ≥75 kg, respectively. Peginterferon-alfa 2a 180 μg was administered once weekly via subcutaneous injection. This was an open-label study, treatment assignments were not masked to patients or study administrators or investigators at any point.
The use of erythropoietin-stimulating agents, granulocyte colony-stimulating factors, or platelet boosters was prohibited from 28 days before baseline through the end of treatment.
Assessments
Samples for determining plasma HCV RNA levels were drawn at screening, on-treatment time points, including day 1, weeks 1, 2, 4, 8, 10, 12, and, where applicable, 16, 20, and 24, and at post-treatment weeks 4, 12, and 24. Plasma HCV RNA was analyzed using the Roche Cobas TaqMan HCV RNA CAP/CTM Test, v2.0 for use with the High Pure System (Roche Molecular Systems, Inc., Branchburg, NJ), which has a lower limit of quantification (LLOQ) of 15 IU/mL.
For analysis of viral resistance, we collected blood samples before dosing (baseline) and at each subsequent visit for all patients. For patients with virologic failure, we compared samples taken at baseline and time of failure to detect any amino acid changes in the nonstructural 5B polymerase region that might confer resistance to sofosbuvir. We report resistance-associated variants (RAVs) that were present in at least 1% of deep sequence reads.
Safety data collected during treatment included reported adverse events, physical examinations, clinical laboratory tests, vital signs, and electrocardiography recordings. Concomitant medication intake was also recorded. Treatment-emergent clinical and laboratory adverse events were summarized using the Medical Dictionary for Regulatory Activities (version 17.1).
End Points and Statistical Analyses
The primary efficacy end point was the percentage of patients in each study group with SVR12, defined as HCV RNA less than LLOQ (15 IU/mL) 12 weeks after stopping the study drug. For the primary efficacy analysis, the SVR12 rate was calculated with a 2-sided 95% exact confidence interval using the Clopper-Pearson method. Patients without HCV RNA result for post-treatment week 12 for any reason were counted as treatment failures.
The primary statistical hypothesis testing was performed in all patients with genotype 3 HCV who were enrolled and received at least one dose of study treatment. A sequence of noninferiority/superiority hypotheses comparing the treatment groups was conducted using a gatekeeping approach to control the familywise error rate. The comparison of the SVR12 rates between any 2 treatment groups was performed using a Cochran-Mantel-Haenszel test stratified by prior treatment experience and presence/absence of cirrhosis. In stage 1 of the analysis, we assessed the equivalence of the SVR12 rates for patients receiving 16 or 24 weeks of sofosbuvir and ribavirin. Equivalence was evaluated using a two 1-sided testing approach. The hypotheses at later stages were defined separately, depending on the outcomes of stage 1. See Supplementary Material for the sequence of hypotheses. The SVR12 rate of one group was considered superior to that of another group if the P value of the superiority test (Cochran-Mantel-Haenszel test) was less than the α levels prespecified (α = .025 one-sided for 24 weeks compared with 16 weeks and 12 weeks compared with 16 weeks, and α = .0125 one-sided for 12 weeks compared with 24 weeks) (Supplementary Material).
Secondary efficacy end points included the percentage of patients in each group with HCV RNA less than LLOQ during treatment and rates of relapse and breakthrough.
We calculated that a sample size of 172 subjects per arm would provide >83% power to detect a difference of 15% in SVR12 rates between 16 and 24 weeks of sofosbuvir plus ribavirin at a .0125 significance level using a 1-sided continuity-corrected χ2 test. Assuming an SVR12 rate of 85% for the groups receiving 24 weeks of sofosbuvir and ribavirin and 12 weeks of sofosbuvir plus peginterferon and ribavirin, this sample size would provide >80% power to establish noninferiority of 24 weeks of sofosbuvir plus ribavirin to 12 weeks of sofosbuvir plus peginterferon and ribavirin (with a noninferiority margin of 12%) at a signicance level of .0125 using a 1-sided test.
Role of the Funding Source
The sponsor of the study designed and undertook the study, and collected and analyzed the data, in collaboration with external investigators. The sponsor and Graham R. Foster interpreted data. The manuscript was drafted by Jennifer King of August Editorial, who was paid by the sponsor. All authors had full access to study data and approved the final manuscript.
Results
Baseline Characteristics
We screened 776 patients, of which 601 were randomized, and 592 received at least one dose of study treatment (Supplementary Material). The baseline demographic and disease characteristics of the patients in the 3 treatment groups are shown in Table 1. A majority of patients were white (84%) and 67% were male. Forty-eight (8%) patients had genotype 2 HCV and 544 (92%) had genotype 3 HCV (of the 522 patients with genotype 3 HCV who could be subtyped, 512 [98%] had genotype 3a HCV). Overall, 37% of patients (100% of those with genotype 2 HCV and 31% of those with genotype 3 HCV) had compensated cirrhosis. Mean baseline HCV RNA levels were similar among the treatment groups.
Patients With Genotype 2 Hepatitis C Virus
Among treatment-experienced patients with genotype 2 HCV and cirrhosis, reduction in viral load to less than LLOQ was markedly more rapid in those receiving peginterferon than in those receiving sofosbuvir and ribavirin: 88% of patients (14 of 16) receiving sofosbuvir plus peginterferon and ribavirin had HCV RNA less than LLOQ by week 2, as compared with 40% of patients (6 of 15) in the 16-week group and 41% (7 of 17) in the 24-week group. By week 8 of treatment, all 48 patients had HCV RNA less than LLOQ.
Rates of SVR12 were similar across treatment groups: 87% of patients (13 of 15) who received 16 weeks of sofosbuvir and ribavirin, 100% of patients (17 of 17) who received 24 weeks of sofosbuvir and ribavirin, and 94% of patients (15 of 16) who received 12 weeks of sofosbuvir plus peginterferon and ribavirin. Three patients with genotype 2 HCV did not achieve SVR12, two patients receiving 16 weeks of sofosbuvir and ribavirin relapsed by post-treatment week 4, and 1 patient receiving sofosbuvir plus peginterferon and ribavirin was lost to follow-up after having HCV RNA less than LLOQ at post-treatment week 4. Deep sequencing in the 2 relapsers did not reveal the sofosbuvir RAVs S282T, L159F, or V321A, at baseline or time of virologic failure.
Patients With Genotype 3 Hepatitis C Virus
Among patients with genotype 3 HCV, on-treatment response was also more rapid in those receiving peginterferon than in those receiving sofosbuvir and ribavirin. By week 2 of treatment, 65% of patients with genotype 3 HCV receiving sofosbuvir plus peginterferon and ribavirin had HCV RNA less than LLOQ, as compared with 54% and 55%, respectively, for genotype 3 patients receiving sofosbuvir and ribavirin for 16 and 24 weeks (P = .007 for both comparisons). By week 8 of treatment, virtually all evaluable patients (99%) had HCV RNA less than LLOQ.
Among patients with genotype 3 HCV, overall rates of SVR12 were 71% in those who received 16 weeks of sofosbuvir and ribavirin, 84% in those who received 24 weeks of sofosbuvir and ribavirin, and 93% in those who received 12 weeks of sofosbuvir plus peginterferon and ribavirin (Table 2). The SVR12 rate among patients who received 12 weeks of sofosbuvir plus peginterferon and ribavirin was statistically superior to that among patients who received 16 weeks of sofosbuvir and ribavirin (by 22 percentage points; 95% confidence interval: 15%-30%; P < .001) and to that of patients who received 24 weeks of sofosbuvir and ribavirin (by 9 percentage points; 95% confidence interval: 2%-16%; P = .008). In addition, the SVR12 rate among patients who received 24 weeks of sofosbuvir and ribavirin was statistically superior to that among those who received 16 weeks of sofosbuvir and ribavirin (by 13 percentage points; 95% confidence interval: 5%-22%; P = .002).
The same pattern of response-the lowest rates of SVR12 among patients receiving 16 weeks of sofosbuvir and ribavirin and the highest among patients receiving 12 weeks of sofosbuvir plus peginterferon and ribavirin-was seen in every major subgroup of patients with genotype 3 HCV, including treatment-naïve and treatment-experienced patients, those with and without cirrhosis, and in subgroups by combined treatment history and cirrhosis status (Table 2, Figure 1, and Supplementary Material).
Among patients with genotype 3 HCV, 83 had virologic relapse after the end of treatment-50 (28%) of those receiving 16 weeks of sofosbuvir and ribavirin, 24 (13%) of those receiving 24 weeks of sofosbuvir and ribavirin, and 9 (5%) of those receiving 12 weeks of sofosbuvir plus peginterferon and ribavirin. We performed an exploratory multivariable logistic regression analysis using the forward selection method to discover baseline factors associated with relapse among patients with genotype 3 HCV. Our results showed that male sex and non-CC allele of the IL28B gene were independently associated with relapse among patients receiving 16 weeks and 24 weeks of sofosbuvir and ribavirin (Table 3). The presence of cirrhosis was independently associated with relapse after treatment with 16 weeks of sofosbuvir and ribavirin and 12 weeks of sofosbuvir plus peginterferon and ribavirin, but not 24 weeks of sofosbuvir and ribavirin.
Among patients receiving sofosbuvir and ribavirin alone, on-treatment response was somewhat slower in those who relapsed than in those who achieved SVR12.
Among those who relapsed, 38% of patients receiving 16 weeks of sofosbuvir and ribavirin and 33% of those receiving 24 weeks of sofosbuvir and ribavirin had HCV RNA less than LLOQ at week 2 of treatment, as compared with 61% (P = .007) and 58% (P = .028) of those who achieved SVR12. By week 4 of treatment, this difference was not as marked (Supplementary Table 10). Among patients receiving sofosbuvir plus peginterferon and ribavirin, on-treatment response among those who relapsed and those who achieved SVR12 was similar.
Deep sequencing of the NS5B polymerase region of the HCV RNA was attempted at baseline and at time of virologic failure in 83 patients with genotype 3 HCV who relapsed. Full-length NS5B sequencing was successful for 81 of the 83 patients at baseline, and from 76 of 83 at time of relapse. The S282T RAV was not present in any patient at any time point. The L159F RAV was identified at levels ranging from 1.2% to >99% of viral sequence reads in 8 patients at the time of relapse, including 1 who had the variant at baseline. The V321A RAV was identified in 1 patient (in 35.1% of viral sequence reads) at the time of virologic failure. One patient who had a low level of V321A (3.8%) at baseline relapsed after treatment, but amplification of NS5B at the time of relapse was not successful. All patients with L159F and V321A at time of relapse were in the sofosbuvir and ribavirin arms. None of the patients receiving sofosbuvir plus peginterferon and ribavirin who were successfully sequenced after virologic relapse had S282T, L159F, or V321A RAVs.
Patients With On-Treatment Virologic Failure
Three patients, all with genotype 3a HCV and all in the group receiving 24 weeks of sofosbuvir and ribavirin, experienced virologic failure during treatment. Two of these patients, both 41-year-old white males, experienced virologic breakthrough. One had HCV RNA less than LLOQ from week 2 through week 8 of treatment, but had quantifiable HCV RNA at the treatment week 12 visit; no additional data are available because the patient was subsequently lost to follow-up after week 12. The other patient with virologic breakthrough had HCV RNA less than LLOQ for the first time at the week 8 visit, but had quantifiable HCV RNA at the following visit (week 12) and at all other visits through post-treatment week 12. The third patient with on-treatment virologic failure, a 50-year-old Asian male, failed to achieve HCV RNA less than LLOQ through week 8 and was therefore discontinued from treatment, after which the patient withdrew consent. Pharmacokinetic analyses of all 3 patients showed detectable plasma levels of sofosbuvir and its metabolites GS-331007 and GS-566500, consistent with drug intake at the scheduled visits. Although drug levels in these 3 patients' results do not indicate lack of adherence, pill counts suggest that 1 patient with virologic breakthrough might not have been adherent. The S282T, L159F, or V321A RAVs were not detected at baseline in any of the 3 patients with on-treatment virologic failure. Amplification was possible for samples taken at the time of virologic failure from 2 of these 3 patients. One was a partial responder who had no RAVs at the time of virologic failure at week 2. In the other patient, who had breakthrough at week 12, the V321A variant was detected at a low level (2.5%).
Safety
The majority of patients experienced at least one adverse event during treatment (Table 4), but most adverse events were mild to moderate in severity. The most common adverse events in all 3 treatment groups were fatigue, headache, and insomnia. Numerically greater proportions of patients in the peginterferon-containing group than in the sofosbuvir and ribavirin-only groups experienced fatigue, nausea, rash, decreased appetite, myalgia, cough, influenza-like illness, pyrexia, and chills. Serious adverse events were experienced at similar frequencies across treatment arms. The proportion of patients with moderate (grade 3) and severe (grade 4) adverse events was not substantially higher in the peginterferon-containing arm (8%) than in the interferon-free arms (6% and 4%, respectively, in patients receiving 16 and 24 weeks of sofosbuvir and ribavirin).
Six (1%) patients, 5 receiving sofosbuvir and ribavirin and 1 receiving sofosbuvir plus peginterferon and ribavirin, discontinued all study treatment because of an adverse event. Three of these discontinuations were not thought to be related to study drugs. Of the 3 discontinuations thought to be related to study treatment, 2 were in patients receiving 16 weeks of sofosbuvir and ribavirin (sleep disturbances in 1 patient and worsening depression/suicidal thoughts in the other) and 1 in a patient receiving sofosbuvir plus peginterferon and ribavirin (depression). Two of these 6 patients achieved SVR12, 2 relapsed, and 2 discontinued the study early.
A numerically greater proportion of patients receiving sofosbuvir plus peginterferon and ribavirin for 12 weeks had grade 3 and 4 laboratory abnormalities than patients receiving sofosbuvir and ribavirin for 16 or 24 weeks. The most common grade 3 and 4 laboratory abnormalities were hematologic. The median reduction in hemoglobin level at the end of treatment was 2.6 g/dL among patients receiving sofosbuvir plus peginterferon and ribavirin, as compared with 2.0 g/dL and 1.8 g/dL, respectively, among patients receiving sofosbuvir and ribavirin for 16 and 24 weeks. Two patients, both in the peginterferon-containing group, experienced declines in hemoglobin to <8.5 g/dL. Four patients, 3 receiving sofosbuvir plus peginterferon and ribavirin and 1 sofosbuvir-ribavirin had blood transfusions (2 in the post-treatment period due to gynecologic bleeding in 1 patient randomized to sofosbuvir plus peginterferon and ribavirin, and the other in a diabetic patient randomized to sofosbuvir and ribavirin who developed pneumonia associated with urinary tract infection). Other grade 3 and 4 cytopenias were more common among patients in the peginterferon-containing arm than among patients receiving sofosbuvir and ribavirin (Supplementary Material).
Discussion
The results of this phase 3 trial have shown that patients with genotype 3 HCV achieve superior rates of SVR12 with 12 weeks of sofosbuvir plus peginterferon and ribavirin than they do with 16 or 24 weeks of sofosbuvir and ribavirin. Numerically superior SVR 12 rates were observed across all major subgroups of genotype 3 patients who received triple therapy as compared with those receiving IFN-free treatment. Additionally, genotype 2 treatment-experienced HCV-infected patients with cirrhosis had high SVR rates in all treatment groups.
For patients with genotype 3 HCV, current treatment guidelines suggest the following options: 24 weeks of sofosbuvir and ribavirin, 12 weeks of sofosbuvir plus peginterferon and ribavirin, or, in Europe, sofosbuvir and daclatasvir for 12 weeks in treatment-naïve or peginterferon-experienced patients, and sofosbuvir and daclatasvir plus ribavirin for 12 or 24 weeks in treatment-experienced patients.1, 2 The first option was based on results of the VALENCE trial, in which 85% (213 of 250) of treatment-naïve and previously treated patients achieved SVR12 after 24 weeks of sofosbuvir and ribavirin.8 Among treatment-experienced patients with cirrhosis, the SVR12 rate was only 62%. The second treatment option is based on results from a small cohort in the phase 2 LONESTAR-2 trial.11 Of the 24 patients with genotype 3 who received sofosbuvir plus peginterferon and ribavirin, 20 (83%) achieved SVR12. In this trial, there was no difference in response between patients with and without cirrhosis. The third and fourth option is based on results from the ALLY-3 trial, in which 90% (91 of 101) of treatment-naïve patients and 86% (44 of 51) of treatment-experienced patients achieved SVR12.9 The SVR12 rate in this trial was substantially lower among patients with cirrhosis receiving 12 weeks of treatment without ribavirin.
Most patients with genotype 2 HCV in the VALENCE trial8 were able to achieve SVR12 with 12 weeks of sofosbuvir and ribavirin. However, a small subgroup (n = 9) of treatment-experienced patients with compensated cirrhosis had an SVR12 rate of 78%, which was considerably lower than rates seen in treatment-naïve and treatment-experienced noncirrhotic patients. In the LONESTAR-2 trial,11 a small subgroup (n = 14) of treatment-experienced genotype 2 patients with cirrhosis had an SVR12 rate of 93% after treatment with 12 weeks of sofosbuvir plus peginterferon and ribavirin. On the basis of these data, current treatment guidelines for patients with genotype 2 HCV who failed previous treatment with peginterferon and ribavirin include, among other options, 16-20 weeks of sofosbuvir and ribavirin or 12 weeks of sofosbuvir plus peginterferon and ribavirin. Our results, albeit in a small group of patients, provide further support for these recommendations, but raise a new question about the optimal duration of sofosbuvir and ribavirin for these patients. Two patients in the 16-week arm relapsed after the end of treatment as compared with no patients in the other 2 treatment groups. Although this suggests that 24 weeks of sofosbuvir and ribavirin is optimal, our study was not powered for formal comparisons among patients with genotype 2 HCV.
On-treatment virologic failures have been reported rarely in clinical trials of sofosbuvir. Most cases on record have been attributable to nonadherence to study drug dosing by patients. In our study, there was no evidence for nonadherence, as the study drug and its metabolites were detectable in the plasma in samples taken during study visits.
In this study, the S282T variant of the NS5B protein-the first identified sofosbuvir RAV-was not observed in any patient at any time point. A recent analysis of 282 patients who did not achieve SVR in phase 2 and 3 studies with sofosbuvir identified 2 additional variants, L159F and V321A, that confer a slightly reduced susceptibility to sofosbuvir.12 In our study, L159F and V321A were observed in a subset of patients with virologic failure, confirming their association with sofosbuvir treatment. All patients with L159F and V321A at the time of relapse were in the sofosbuvir and ribavirin arms, suggesting that the addition of peginterferon to sofosbuvir and ribavirin reduces the emergence of these variants.
The well-known side effects of interferon have made its elimination from the treatment of HCV a primary clinical and research objective.13, 14, 15 One of the main reasons that interferon was so difficult for patients to tolerate was the length of treatment-most interferon-based regimens had durations of 6 months to 1 year, at the end of which patients were faced with a relatively high likelihood of relapse. With a shorter duration of 12 weeks and greater efficacy, the side effects of interferon have proven to be less formidable.5 In our study, the safety profile of the 12-week regimen of sofosbuvir plus peginterferon and ribavirin was unexpectedly not markedly different from that of 16-24 weeks of sofosbuvir and ribavirin. Some interferon-associated side effects were more common in patients receiving this drug (fatigue, nausea, decreased appetite, myalgia, flu-like illness, and rash), but these were typically mild and the rates of adverse events leading to discontinuation of the study drug were similar in all arms. Safety and tolerability did not differ by treatment experience; similar rates of adverse events, serious adverse events, and treatment discontinuation for adverse events were observed in treatment-naïve and previously treated patients (Supplementary Material).
The results of this study are limited by the small number of patients with genotype 2 HCV, which tempers any conclusions about differences between SVR12 rates by regimen in patients with genotype 2 HCV. The study was also not powered for comparisons among subgroups of patients with genotype 3 HCV. However, our results were generally in keeping with those previously reported in phase 2 and 3 trials. Other features of the study population-its covering multiple sites in different continents, and the inclusion of a large proportion of cirrhotic patients with minimum platelets cell counts lower than that usually recommended for interferon-based treatment-support the generalizability of the overall efficacy and safety results to a broad population of patients. The open-label design of the study is unlikely to have biased the reporting of efficacy results because HCV RNA is an objective laboratory assessment, but bias in the reporting of safety events cannot be ruled out. Although the field of HCV treatment is moving rapidly away from interferon-based regimens, our results suggest that a short course of highly effective interferon-containing treatment may still have a role in subpopulations without better options. In particular, SVR rates in treatment-experienced patients with genotype 3 HCV and cirrhosis receiving all-oral combinations-sofosbuvir plus daclatasvir, sofosbuvir plus ledipasvir, and sofosbuvir plus ribavirin-range from 62% to 73%.8, 9, 10
In conclusion, our results provide clear evidence that sofosbuvir plus peginterferon and ribavirin for 12 weeks should continue to be considered a treatment option for eligible patients with genotype 3 HCV. Our results support the use of sofosbuvir and ribavirin for 24 weeks as an option for patients who cannot take interferon or are unwilling to do so.
|
|
| |
| |
|
|
|