| |
TLR7 Nature Immunology research paper & Commentary
|
| |
| |
Download the PDF here
Download the PDF here
CROI: TLR7 Agonist GS-9620 Activates HIV-1 in PBMCs from HIV-Infected Patients on cART - (02/26/15)
CROI: Treatment with a TLR7 Agonist Induces Transient Viremia in SIV-Infected ART-Suppressed Monkeys - (02/27/15)
GS-9620, an Oral Agonist of Toll-Like Receptor-7, Induces Prolonged Suppression of Hepatitis B Virus (and HCV) in Chronically Infected Chimpanzees ......http://www.natap.org/2013/HBV/021913_02.htm
Targeting Innate HBV Immunity: A New Step in the Development of Combination Therapy for Chronic Hepatitis B; GS-9620, an oral agonist of Toll-like receptor-7, induces prolonged suppression of hepatitis B virus in chronically infected chimpanzees......http://www.natap.org/2013/HBV/062613_01.htm
----------------------
2 Nature articles on TLR7 below (including-original research article discussed by Michael Lederman "TLR7 induces anergy in human CD4+ T cells") .........excerpts from original research article-
"Toll-like receptors (TLRs) represent the major pathway by which microorganisms interact with host cells.
......The recognition of microbial patterns by Toll-like receptors (TLRs) is critical for activation of the innate immune system. Although TLRs are expressed by human CD4+ T cells, their function is not well understood. Here we found that engagement of TLR7 in CD4+ T cells induced intracellular calcium flux with activation of an anergic gene-expression program dependent on the transcription factor NFATc2, as well as unresponsiveness of T cells. As chronic infection with RNA viruses such as human immunodeficiency virus type 1 (HIV-1) induces profound dysfunction of CD4+ T cells, we investigated the role of TLR7-induced anergy in HIV-1 infection. Silencing of TLR7 markedly decreased the frequency of HIV-1-infected CD4+ T cells and restored the responsiveness of those HIV-1+ CD4+ T cells. Our results elucidate a previously unknown function for microbial pattern-recognition receptors in the downregulation of immune responses.......Treatment with the synthetic TLR7 agonist imiquimod resulted in considerably less proliferation of CD4+ T cells than that of vehicle-treated control cells, as well as less secretion of IFN-γ and IL-17, in a dose-dependent fashion (Fig. 1c-e and Supplementary Fig. 1c,d). We observed this inhibitory effect as soon as 12 h after activation, with much less induction of the expression of IL2, IFNG and IL4 after treatment with imiquimod (Supplementary Fig. 1e)......Here we have described a previously unknown function of these pattern-recognition receptors in shutting off the immune responses of human CD4+ T cells. Engagement of TLR7 by its ligands in CD4+ T cells prevented entry into the cell cycle and the secretion of proinflammatory cytokines after stimulation.......engagement of TLR7 by its ligand is sufficient to induce activation and secretion of proinflammatory cytokines in monocytes, as expected for a cell of the innate immune system.......Together these results showed that inhibition of TLR7 signaling was able to diminish the HIV-1 load in infected CD4+ T cells derived from infected patients......We note that all the ligands we used here were pharmacological agonists of TLR7. We have not been able to find any immunostimulatory RNA specific for human TLR7, which would be a more physiological ligand, as these immunostimulatory RNA sequences are specific for mouse TLR7 but recognize human TLR8 instead. We have overcome this issue through the use of HIV-1 as a potential 'natural' ligand of TLR7. The physiological relevance of our data is suggested by the effect of TLR7 signaling on HIV-1-infected cells......the observations reported here suggest involvement of TLR7 both in the induction of the anergic phenotype observed in HIV-1-infected cells and in increases in the degree of infection of CD4+ T cells, which suggests that TLR7-induced upregulation of calcium is involved in the viral life cycle.....In summary, we have demonstrated a previously unknown role for TLR7 in CD4+ T cell function that is in direct opposition to its role in cells of the innate immune system. Moreover, TLR7 ligands may be used as a means of inducing 'tolerance' on CD4+ T cells in human autoimmune diseases. Finally, our data have demonstrated a novel function for microbial pattern-recognition receptors in the inhibition of immune responses."
--------------------
A surprising role for TLR7
Michael M Lederman
Nature Immunology 16, 8-9 (2015)
Ligation of the Toll-like receptor TLR7 in human CD4+ T cells elicits an anergic state that may contribute to CD4+ T cell hyporesponsiveness after infection with human immunodeficiency virus type 1 and may also enhance propagation of this virus.
TLR7 is one of several Toll-like receptors (TLR3, TLR7, TLR8 and TLR9) that recognize microbial nucleic acid sequences. TLR7 and TLR8, which recognize single-stranded RNA, are distributed broadly among myeloid and other cells and are characteristically expressed in endosomal compartments, where their engagement with microbial sequences is thought to take place. In this issue of Nature Immunology, Dominguez-Villar et al. demonstrate a surprising role for TLR7 in host-virus relationships1. In a paper fraught with novelty, the authors provide evidence that engagement of TLR7 expressed in primary human CD4+ T cells can induce anergy. They also find that activation of a TLR7-dependent mechanism in CD4+ T cells promotes the replication of human immunodeficiency virus type 1 (HIV-1) within these cells-a hitherto unexpected role for this microbial sensor.
The authors first report that incubation of human CD4+ T cells with various TLR7 agonists such as imiquimod impairs their proliferation and the expression of various cytokines in response to costimulation via the invariant signaling protein CD3 and coreceptor CD28. They do not obtain a similar effect with TLR8 agonists, and this effect of TLR7 stimulation is abrogated by silencing of TLR7-encoding RNA; therefore, the proanergic effect seems to be a specific property of TLR7 ligation. There is an emerging literature suggesting that non-synchronized activation of the T cell antigen receptor and costimulation results in calcium flux and induction of anergy through activation of the transcription factor NFAT1 (ref. 2). Dominguez-Villar et al. now show that engagement of TLR7 in CD4+ T cells results in induction of calcium flux and activation of the transcription factor NFATc2 via dephosphorylation that then drives the expression of a host of anergy-related genes1. This effect is abrogated by silencing of the gene encoding NFATc2. The engagement of TLR7 with imiquimod also results in phosphorylation of the transcription factor NF-κB and the signaling molecules IRAK4 and p38 but lower basal expression of the signaling molecule Jnk and less phosphorylation of Jnk and its target Jun (a component of the transcription factor AP-1) induced by the phorbol ester PMA plus ionomycin. Collectively these results offer two potential means by which stimulation via TLR7 diminishes the immunological responsiveness of CD4+ T cells: one by the induction of various anergy-related genes, and the other by interference with AP-1-dependent signals.
To try to place their findings into a more physiological context, the authors seek to understand whether the hyporesponsive state of CD4+ T cells seen in HIV infection could be explained by that TLR7 effect. The authors confirm earlier reports that infection of CD4+ T cells with HIV-1 in vitro diminishes their ability to produce the cytokines IL-2 and IFN-γ after restimulation. Productive infection is apparently not necessary for this anergic effect, as cells that have not supported expression of an HIV-1 reporter gene seem to be as compromised in these assays as are their neighboring productively infected cells. It might be a stretch to contend that this phenomenon drives the dysfunction of CD4+ T cells in untreated HIV infection, as an even smaller minority of CD4+ T cells seem to be HIV-1 infected in vivo. However, in the setting of unrestrained viral infection that is linked to both clinical evidence and laboratory evidence of CD4+ T cell dysfunction, it may be possible that many CD4+ T cells are in fact exposed to virus, viral proteins or viral sequences that do not reflect completion of the viral life cycle but still may engage the coreceptor CD4, TLR7 or other innate sensors3.
The authors then report an unexpected role for engagement of TLR7 that facilitates the infection of CD4+ T cells with HIV-1, as silencing of the gene encoding TLR7 or blockade of this innate receptor with the inhibitor IRS661 attenuates infection. Here too a role for NFATc2 is suggested, as silencing of the gene encoding NFATc2 or blockade of NFATc2 itself also attenuates HIV-1 replication. The authors conclude that HIV-1 uses the anergic state induced by ligation of TLR7 to support its own propagation. As best as can be seen, their systems use broad activation of T cells to render target cells susceptible to in vitro infection, but still it is fascinating to propose that HIV has 'learned' to utilize for its own propagation in CD4+ T cells an innate sensor that is typically used by dendritic cells, monocytes and macrophages to arm host defenses against viruses. Indeed, the authors find no evidence that engagement of TLR7 on CD14+ monocytes leads to a hyporesponsive state; instead, and as expected, these cells are activated following exposure to imiquimod. Ligation of TLR7 in CD4+ T cells therefore seems to trigger a response entirely different from that seen in monocytes. Why would this be? At this point, researchers can only speculate. Perhaps engagement of TLR7 helps to defend (in myeloid cells, via the activation of antiviral defenses; and in T cells, via the attenuation of their activation) against archaic retroviruses that have successfully populated 7% of the human genome. Moreover, while the activation of T cells drives adaptive immunity, it seems that checks and balances are needed, such that signals are coordinated in place and time (and magnitude) to promote an effective (and tolerated) immune response.
On the other hand, the authors propose that this model of TLR7-dependent anergy has a role in the persistence of HIV and at the level of sustaining virus-infected cells and impairment of adaptive defenses, this may be so, but there are aspects of this story that will need some more sorting out. For example, does the engagement of TLR7 take place after viral entry and 'decapsidation', or do newly transcribed viral RNAs bind TLR7, either way sustaining the anergic state and promoting viral replication (Fig. 1)? Do viral sequences enter TLR7-containing endosomes via autophagy4, or do some viruses enter CD4+ T cells via endosomes5? And at what stage of the viral life cycle does this anergic state enhance viral replication? The methods used here do not distinguish events before integration from those after integration, but the activation of both NFATc2 and NF-κB by ligation of TLR7 is compatible with the idea that engaging TLR7 in CD4+ T cells may enhance transcription from the viral promoter.
Figure 1: The engagement of TLR7 in isolated CD4+ T cells induces an anergic state and promotes HIV-1 replication.
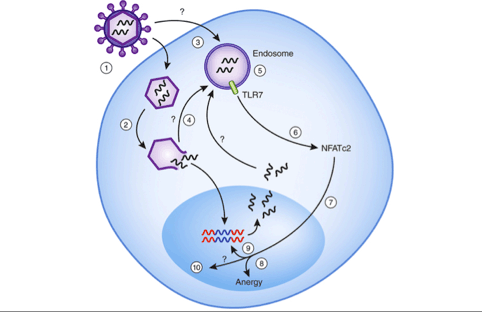
HIV enters CD4+ T lymphocytes after fusion with the cell membrane (1). As the capsid decomposes, viral RNA is reverse-transcribed and proviral DNA then enters the nucleus and integrates into the host genome (2). Potentially, some virions could enter the cell via endosomes (3); alternatively, during decapsidation, some virions might enter phagosomes via autophagy (4). Within endosomes, TLR7 is engaged by viral single-stranded RNA, which results in calcium flux (5). The calcium flux results in dephosphorylation of NFATc2 (6), which translocates to the nucleus (7), where a series of anergy-related genes are induced (8). Activated NFATc2 (and NF-κB that is activated by ligation of TLR7) may also enhance the transcription of viral RNA from integrated HIV (9) or could potentially induce expression of other host factors that promote the HIV replication cycle at sites to be determined (10).
Marina Corral Spence/Nature Publishing Group
However, in myeloid cells, ligation of TLR7 typically initiates an antiviral state by driving expression of type 1 interferons that arm global antiviral defenses and also induce expression of various elements that restrict HIV replication directly6. In this context, it remains unknown whether TLR7 ligation or HIV replication within these CD4+ T cells activates a type I interferon response and whether the immunosuppressive effects of type I interferons acting in trans contribute to the broad impairment of immunological function seen here even in cells that are not HIV-1 infected.
Is TLR7-induced anergy a prerequisite for HIV-1 infection? Perhaps in this system it is, but it may be overreaching to conclude that this phenomenon is central to HIV propagation, as both here and in other laboratory settings, activation of CD4+ T cells is used to render cells more readily infectible, and in clinical settings, activation of the immune system is linked to HIV replication both as its consequence and also as its plausible driver7. However, maybe there is some warning to 'lumpers' (like me) that the relationship between immunity and viral propagation in HIV infection might be more nuanced. Indeed, current fashion dictates that HIV 'likes' activation of the immune system, replicating better in activated cells than in resting cells, and somehow drives activation of the immune system, pathogenesis and morbidity. However, at the same time, people with HIV infection can have a profoundly impaired immune system, with not only decreased numbers of circulating CD4+ T cells but also a functional anergy-like impairment of the cells that remain. On the other hand, it is possible that the systems applied to test models in vitro introduce more paths and diversions than are justified by whole-person biology. Nonetheless, this is a lovely, clearly presented work that will undoubtedly stimulate more of the same, plenty of arguments and maybe even the hurling of some fruit. Hurrah!
------------------------------
TLR7 induces anergy in human CD4+ T cells
Margarita Dominguez-Villar1, Anne-Sophie Gautron1, Marine de Marcken1, Marla J Keller2 & David A Hafler1
Nature Immunology 16, 118-128 (2015)
The recognition of microbial patterns by Toll-like receptors (TLRs) is critical for activation of the innate immune system. Although TLRs are expressed by human CD4+ T cells, their function is not well understood. Here we found that engagement of TLR7 in CD4+ T cells induced intracellular calcium flux with activation of an anergic gene-expression program dependent on the transcription factor NFATc2, as well as unresponsiveness of T cells. As chronic infection with RNA viruses such as human immunodeficiency virus type 1 (HIV-1) induces profound dysfunction of CD4+ T cells, we investigated the role of TLR7-induced anergy in HIV-1 infection. Silencing of TLR7 markedly decreased the frequency of HIV-1-infected CD4+ T cells and restored the responsiveness of those HIV-1+ CD4+ T cells. Our results elucidate a previously unknown function for microbial pattern-recognition receptors in the downregulation of immune responses.
Introduction
Toll-like receptors (TLRs) represent the major pathway by which microorganisms interact with host cells. They are a family of highly conserved pattern-recognition receptors that recognize distinct pathogen-associated molecular patterns that are conserved in specific classes of microorganisms1. The human TLR family consists of at least ten members that can be classified into two different groups on the basis of their cellular location. Intracellular TLRs (TLR3, TLR7, TLR8 and TLR9) recognize nucleic acids; TLR7 and TLR8 recognize single-stranded RNA2, 3, whereas TLR3 and TLR9 are receptors for double-stranded RNA and double-stranded DNA, respectively. In contrast, cell surface TLRs (TLR1, TLR2, TLR4, TLR5 and TLR6) recognize various components of bacteria1. In mice, although TLR7 and TLR8 are expressed at low levels in CD4+ T cells, there are species-specific differences in the recognition of ligands3 as well as in their functionality. Specifically, mouse TLR7 and human TLR8 mediate species-specific recognition of GU-rich single-stranded RNA. It has been suggested that in contrast to its human TLR counterpart, mouse TLR8 is not functional and TLR7 is the only TLR that recognizes single-stranded RNA4. The expression and signaling pathways triggered by stimulation of TLRs have been described in antigen-presenting cells (APCs) in a process that leads to the activation of APCs with the secretion of inflammatory and antiviral cytokines1, 5. Although TLR expression has been studied mainly in APCs, several reports have described the expression of TLRs on lymphocytes6, and specifically on CD4+ T cells. As with APCs, such studies indicate that the engagement of TLRs acts as a positive costimulatory signal that increases the secretion of proinflammatory cytokines, proliferation and cell survival7, 8.
While TLRs are central to the early host immune response to acute viral infection, more-chronic infectious diseases are characterized by the inability of the host immune system to mount a strong, long-lasting response to the infectious agent. In particular, it has been shown that during infection with RNA viruses such as hepatitis C virus and human immunodeficiency virus type 1 (HIV-1), the immune responses mediated by CD4+ helper T cells and CD8+ cytotoxic T cells determine the outcome of the infection, with chronic infections being correlated with late, transient or narrowly focused responses by CD4+ or CD8+ T cells9, 10, 11. Several studies have demonstrated impairment in the activation and/or function of T cells during infection with HIV-1. Specifically, CD4+ T cells from patients chronically infected with HIV-1 display an anergic phenotype with defects in proliferation and the secretion of interleukin 2 (IL-2) and interferon-γ (IFN-γ). The mechanisms by which RNA viruses impair T cell function are not well understood.
Here we describe a previously unrecognized pathway of TLR-mediated negative regulation of both the activation and the cytokine production of CD4+ T cells. Engaging TLR7 expressed on CD4+ T cells resulted in complete anergy by inducing intracellular calcium flux, with activation of an anergic gene-expression program dependent on the transcription factor NFATc2 and with subsequent T cell unresponsiveness that was reversed by knockdown of TLR7 with short hairpin RNA (shRNA). In studies of the potential physiological relevance of these findings, we found that knockdown of TLR7 via shRNA decreased the frequency of HIV-1-infected CD4+ T cells in vitro and restored the responsiveness of those HIV-1+ CD4+ T cells in vitro. Our results elucidate a previously unknown function for microbial pattern-recognition receptors in the downregulation of immune responses, inducing anergy by increasing intracellular calcium concentrations and interfering with secondary costimulation signals in the presence of signaling via TLRs7.
Results
Inhibition of the activation of CD4+ T cells by TLR7
While investigating a potential costimulatory role for TLRs in CD4+ T cells, we observed that the entry of CD4+ T cells into the cell cycle after crosslinking of the T cell antigen receptor (TCR) with antibody to the invariant signaling protein CD3 (anti-CD3) and crosslinking of the coreceptor CD28 with anti-CD28 was blocked by coengagement of TLR7 (Fig. 1a,b and Supplementary Fig. 1a,b). Treatment with the synthetic TLR7 agonist imiquimod resulted in considerably less proliferation of CD4+ T cells than that of vehicle-treated control cells, as well as less secretion of IFN-γ and IL-17, in a dose-dependent fashion (Fig. 1c-e and Supplementary Fig. 1c,d). We observed this inhibitory effect as soon as 12 h after activation, with much less induction of the expression of IL2, IFNG and IL4 after treatment with imiquimod (Supplementary Fig. 1e). We assessed the effect of concentrations of up to 15 μg/ml of imiquimod but found no effect on cell viability (data not shown). The diminished proliferation correlated with less secretion of the cytokines IFN-γ, IL-17, IL-2 and IL-4, as measured by enzyme-linked immunosorbent assay (ELISA), at day 3 after stimulation (Fig. 1c). We confirmed the diminished cytokine secretion at the single-cell level, as the frequency of cytokine-producing cells was also diminished in a dose-dependent manner with increasing doses of imiquimod in culture (Fig. 1d,e). Furthermore, stimulation of CD4+ T cells in the presence of imiquimod inhibited the expression of activation markers such as CD25, CD69 and CD137, measured 48 h after activation (Fig. 1f). Of note, the effect of imiquimod was not related to the conversion of CD4+ T cells into a population of regulatory T cells (Treg cells), as neither expression of the transcription factor Foxp3 nor secretion of IL-10 was increased in the presence of imiquimod (data not shown). The unresponsive phenotype observed was not due to an indirect effect of TLR7 on Treg cells, as we obtained the same results with a CD4+ T cell population sorted as CD3+CD4+ or CD4+CD25loCD127+ cells and depleted of Treg cells (data not shown). Only stimulation of TLR9 with the synthetic ligand oligodeoxynucleotide ODN2006 also decreased the frequency of proliferating CD4+ T cells, but it did so to a lesser extent than did stimulation of TLR7 (Supplementary Fig. 1a,b). The effect observed was not due to an indirect effect of the triggering of TLR7 on contaminant APCs in the culture, as imiquimod exerted the same effect on T cell clones grown from a single donor (Supplementary Fig. 2).
To confirm the specificity of our results, we stimulated CD4+ T cells in the presence of other synthetic ligands of TLR7, such as loxoribine, CL264 or gardiquimod (Fig. 2). The three ligands tested induced a significantly less CD4+ T cell proliferation than did a vehicle control, with gardiquimod and loxoribine inhibiting proliferation to a degree similar to that achieved by imiquimod treatment (Fig. 2a,b). The secretion of IL-2, IFN-γ, IL-4 and IL-17 was also inhibited after stimulation in the presence of each of the three ligands, as measured by ELISA after 3 d in culture (Fig. 2c). The specificity of the phenotype observed was further demonstrated by silencing of TLR7 in CD4+ T cells (Fig. 2d). While control CD4+ T cells transduced with nontargeting shRNA responded to treatment with imiquimod by decreasing their secretion of IFN-γ and IL-2, CD4+ T cells in which TLR7 was silenced that were stimulated with imiquimod produced concentrations of IFN-γ and IL-2 similar to those produced after treatment with vehicle (Fig. 2e). Both TLR7 and TLR8 are expressed in human CD4+ T cell subpopulations6, 8; however, although reports suggest that both TLR7 and TLR8 recognize single-stranded RNA as their natural ligand in APCs2, 3, we observed no such effect when we used the TLR8 ligand ssRNA40 (a single-stranded RNA) in the transfection reagent LyoVec in these experiments in parallel with TLR7 ligands (Fig. 2f,g). Instead, stimulation of T cells in the presence of ssRNA40 significantly increased the production of IFN-γ and inhibited the secretion of IL-4 by CD4+ T cells (Fig. 2g), with no effect on proliferation (Fig. 2f). Vehicle alone (LyoVec) did not have any effect on cytokine secretion or proliferation under these experimental conditions (data not shown). We obtained the same results with CL075, another ligand predominantly of TLR8 (data not shown). These data were in agreement with a published report8 and suggested that despite sharing ligands, the signaling pathways that TLR7 and TLR8 triggered on CD4+ T cells led to different phenotypic outcomes.
Ligation of TLR7 induces the activation and maturation of APCs, with upregulation of activation markers and secretion of proinflammatory cytokines12, 13, 14. In agreement with published data, CD14+ monocytes isolated from the same donors studied above (Figs. 1 and 2) and stimulated in the presence or absence of imiquimod showed an activated phenotype, with upregulation of the expression of the human leukocyte antigen HLA-DR, the costimulatory molecule CD80 and the cytokine receptor CD25 and downregulation of the expression of the costimulatory molecule CD86 (Supplementary Fig. 3a). Upon being stimulated with imiquimod, CD14+ monocytes also secreted the proinflammatory cytokines IL-6, tumor-necrosis factor and IL-1ß and decreased their secretion of IL-10 (Supplementary Fig. 3b), whereas we observed no effect on monocyte proliferation (data not shown); this suggested that stimulation of CD4+ T cells and CD14+ monocytes with imiquimod led to completely different outcomes.
To confirm the specificity of the unresponsive phenotype driven by TLR7 signaling, we stimulated CD4+ T cells with anti-CD3 and anti-CD28 in the presence of shRNA specific for TLR7 or nontargeting shRNA, as a control. After 5 d of culture, we confirmed that the efficiency with which protein and RNA was silenced was >80% (Fig. 2d). We stimulated shRNA-treated resting CD4+ T cells with with anti-CD3 and anti-CD28 in the presence or absence of imiquimod and measured the secretion of IL-2 and IFN-γ. While CD4+ T cells transduced with nontargeting shRNA showed a decrease in the production of IL-2 and IFN-γ secretion after treatment with imiquimod, this effect was abolished in cells expressing TLR7-specific shRNA (Fig. 2e), which confirmed that the unresponsive state of T cells observed in the presence of imiquimod was TLR7 specific.
Induction of calcium-NFATc2-driven CD4+ T cell anergy by TLR7
The inhibition of the proliferation, cytokine secretion and activation of CD4+ T cells upon stimulation in the presence of TLR7 ligands resembled the unresponsive phenotype that characterizes clonal anergy15. Various model systems have been used to induce clonal anergy, including treatment with the calcium ionophore ionomycin16, 17, and the activation of calcium signaling in the absence of activating signals in costimulatory signaling pathways is common in all these models. Thus, the main characteristic of an stimulus that elicits anergy is its ability to induce an unopposed increase in intracellular calcium concentrations17. To test the hypothesis that TLR7 signaling on CD4+ T cells was inducing clonal anergy, we stained sorted CD4+ T cells with the ratiometric calcium indicator Indo-1 AM and treated them with various doses of imiquimod (Fig. 3a). Imiquimod induced a significant and sustained (maintained for at least 20 min) increase in intracellular calcium concentration in a dose-dependent manner that was TLR7 specific, as blocking TLR7 with IRS 661, a specific inhibitory oligonucleotide sequence18, impaired the increase in calcium concentration upon stimulation with imiquimod (Fig. 3b and Supplementary Fig. 4a). We did not observe that increase in intracellular calcium concentration in cells stimulated with the TLR8 agonist ssRNA40 or with ligands of other intracellular TLRs, such as the synthetic RNA duplex poly(I:C) (for TLR3) or ODN2006 (for TLR9) (Fig. 3a and Supplementary Fig. 4b). As a positive control, we treated cells with the calcium ionophore ionomycin, which has been used as an anergy-inducing agent in in vitro experiments17, 19, and obtained concentrations of intracellular calcium similar to those induced by 10 μg/ml of imiquimod (Fig. 3a and Supplementary Fig. 4a).
An immediate consequence of an increase in the concentration of intracellular calcium is the activation of NFATc2, a transcription factor that is highly phosphorylated in resting cells and becomes dephosphorylated by the calcium-calmodulin-dependent phosphatase calcineurin when the concentration of intracellular calcium is increased20, 21. To determine whether stimulation with imiquimod induced dephosphorylation of NFATc2, we stimulated CD4+ T cells with imiquimod and purified total protein extracts 0, 45 and 90 min later. Immunoblot analysis with anti-NFATc2 showed that treatment with imiquimod resulted in dephosphorylation of NFATc2; we confirmed these results by analysis of total extracts of CD4+ T cells stimulated for 40 min with imiquimod and probed with a phosphorylation-specific antibody to NFATc2 (Fig. 3c).
After being dephosphorylated, NFATc2 translocates to the nucleus, where it becomes transcriptionally active22. We used nuclear and cytoplasmic protein extracts to further confirm translocation of NFATc2 from the cytoplasm to the nucleus upon stimulation with imiquimod (data not shown). This translocation of NFATc2 to the nucleus in the absence of a concomitant costimulatory signal leads to the transcription of a set of NFATc2-dependent, anergy-related genes that are different from those upregulated with full activation in the presence of a costimulatory signal17. To investigate whether stimulation of TLR7 and subsequent dephosphorylation of NFATc2 induce the expression of anergy-related genes17, we incubated CD4+ T cells for 2-16 h with imiquimod in the presence or absence of the TLR7-inhibitory sequence IRS 661 and isolated RNA for analysis. As a control for the expression of anergy-related genes, we incubated CD4+ T cells with either ionomycin plus the phorbol ester PMA (a non-anergic stimulus) or ionomycin alone (an anergic stimulus)17 (Supplementary Fig. 5). Ten of the twelve anergy-related genes examined were significantly upregulated in cells stimulated with imiquimod relative to their expression in the control cells stimulated with vehicle (Fig. 3d and Supplementary Fig. 5). The effect observed in the regulation of these genes was TLR7 specific, as preincubation of CD4+ T cells with IRS 661 before treatment with imiquimod abrogated the increase in the expression of anergy-related genes (Fig. 3d). Furthermore, we also assessed the expression of other genes encoding molecules that have been functionally related to establishment and maintenance of the anergic phenotype and are targets of NFAT, such as SIRT1 (ref. 23), ITCH24, CBLB24, 25, DGKA26, EGR2 and EGR3 (ref. 27), and found that all of these but EGR2 showed significant upregulation upon treatment with imiquimod (Fig. 3d). Our data suggested that imiquimod induced clonal anergy in CD4+ T cells via an increase in intracellular calcium concentration and activation of an NFATc2-dependent anergy-related gene-expression program. To further investigate the role of NFATc2 in TLR7-induced T cell anergy, we used two shRNAs to silence NFATC2 and used imiquimod to induce anergy in resting CD4+ T cells after knockdown of NFATc2. After 3 d in culture, cells transduced with nontargeting shRNA and stimulated with imiquimod significantly reduced their production of IL-2 and IFN-γ, while cells transduced with NFATC2-specific shRNA were not affected by treatment with imiquimod (Fig. 3e and Supplementary Fig. 6); this suggested that NFATc2 was necessary for the anergic phenotype driven by TLR7 signaling in CD4+ T cells. Treatment with imiquimod did not have an effect on the expression of NFATC2 (Supplementary Fig. 6a). In agreement with those data and published observations17, treatment with imiquimod failed to upregulate the expression of anergy-related genes (KMD6B, IKZF1, GRG4 and RAB10) in CD4+ T cells in which NFATc2 was knocked down relative to their expression in cells transduced with nontargeting shRNA (Supplementary Fig. 6b), which confirmed that this gene-expression program driven by treatment of CD4+ T cells with imiquimod was largely NFATc2 dependent.
As reported in published studies of an ionomycin-induced anergy model17, we hypothesized that pretreatment of CD4+ T cells with imiquimod would be sufficient to diminish their subsequent cytokine secretion and proliferative response to stimulation with both crosslinking of the TCR and costimulatory signaling through CD28. We pretreated memory CD4+ T cells for 2 h with imiquimod in the presence or absence of the inhibitory sequence IRS 661 and allowed them to 'rest' for 12 h after washout to remove all traces of imiquimod and then stimulated the cells for 3 d with anti-CD3 and anti-CD28. Cells pretreated with imiquimod showed significantly lower production of both IL-2 and IFN-γ than that of cells pretreated with vehicle (Fig. 3f). We observed no significant change in cell viability upon pretreatment wtih imiquimod (data not shown). Again, the pro-anergic effect of imiquimod was prevented by pretreatment with the TLR7 antagonist IRS 661 (Fig. 3f). These data suggested that imiquimod induced clonal anergy in CD4+ T cells by inducing calcium-dependent activation of NFATc2 and subsequent expression of anergy-related genes.
Effect of TLR7 signaling on costimulatory signals
TLR7 signaling has been studied mainly in APCs, in which it is linked to activation of the transcription factors IRF7 and NF-κB and the kinase Jnk through pathways dependent on or independent of the adaptor MyD88 and the kinase IRAK4 (ref. 1). To assess the consequences of TLR7 signaling on CD4+ T cells relative to the consequences of 'conventional' TLR signaling, we isolated CD4+ T cells from healthy donors and stimulated the cells ex vivo with imiquimod, then assessed the phosphorylation status of IRAK4, Jnk, the p65 subunit of NF-κB and the mitogen-activated protein kinase p38 by flow cytometry. In agreement with published reports of other cell types2, 3, 5, stimulation of TLR7 with imiquimod induced phosphorylation of IRAK4, NF-κB and p38 at different time points than did treatment with vehicle (Fig. 4). Preincubation with IRS 661 before stimulation with imiquimod inhibited the phosphorylation of these molecules (Fig. 4), which suggested that the effect observed was TLR7 specific. Incubation with IRS 661 by itself did not have any effect on protein phosphorylation (data not shown). Engagement of TLR7 decreased the basal levels of phosphorylated Jnk (Fig. 4). One of the targets phosphorylated by activated Jnk is Jun, a component of AP-1, which is an essential transcription factor involved in costimulatory signal transduction17. We hypothesized that inhibition of Jnk activity by imiquimod might explain, at least in part, the anergic phenotype observed in CD4+ T cells after stimulation with imiquimod in the presence of full stimulation of the TCR and costimulatory signaling (Figs. 1 and 2). To test this hypothesis, we stimulated CD4+ T cells with PMA plus ionomycin in the presence or absence of imiquimod and assessed the phosphorylation status of Jnk and Jun at various time points. We used NF-κB and p38 as positive controls. Although treatment with imiquimod did not produce an additive effect on the phosphorylation of NF-κB with stimulation, imiquimod further increased the phosphorylation of p38 (Fig. 5a). Of note, upon stimulation with PMA plus ionomycin, the phosphorylation of Jnk was inhibited by imiquimod, and this decrease in Jnk activity was accompanied by a decrease in the phosphorylation of Jun (Fig. 5a). Moreover, imiquimod decreased the activity of both Jnk and Jun, as measured by phosphorylation after activation (Fig. 5a), and it decreased JUN expression (Fig. 5b). These data supported the hypothesis that imiquimod treatment both induced anergy in CD4+ T cells and interfered with costimulatory signaling during T cell stimulation. The decrease in the expression of CD69 and CD137 (both transcriptional targets of AP-1 (refs. 28,29)) observed after stimulation in the presence of imiquimod (Fig. 1f) further supported this hypothesis.
Inhibition of HIV-1 in vitro via TLR7-Ca2+ signaling blockade
To investigate the potential biological and clinical relevance of the observations noted above in ex vivo and in vitro model systems, we determined whether engagement of TLR7 by a single-stranded RNA virus2, 3 could mediate CD4+ T cell anergy. The responses of CD4+ T cells from patients chronically infected with HIV-1 are impaired and are insufficient to clear the virus, while the cells display features of anergy30, 31, 32, 33. Several viral proteins have been shown to induce the state of T cell unresponsiveness that precedes the loss of CD4+ T cells in HIV-1-infected patients30. We hypothesized that a virus-CD4+ T cell interaction through TLR7 would be responsible, at least in part, for the anergic phenotype observed in HIV-1-infected CD4+ T cells from patients. First, we isolated CD4+ T cells from four healthy donors, infected the cells in vitro with physiological concentrations (multiplicity of infection, 0.001) of a replication-competent strain of HIV-1 derived from the prototype HIV-1NL432 virus and tagged with the red fluorescent reporter DsRed (HIV-1NL-D)34, 35 and assessed their ability to produce IL-2 and IFN-γ 7 d after infection. In agreement with published reports30, 31, 32, 33, infection with HIV-1NL-D markedly diminished the ability of viable CD4+ T cells to produce IL-2 and IFN-γ after 4 h of stimulation with PMA and ionomycin (Supplementary Fig. 7). We did not observe this decrease in cytokine secretion only in the bulk T cell population infected with the virus but specifically in HIV-1NL-D-infected cells, which suggested that direct interaction of the virus with infected CD4+ T cells rendered the CD4+ T cells unresponsive.
To test the hypothesis that TLR7 signaling in CD4+ T cells accounted in part for the anergic phenotype after infection with HIV-1, we isolated CD4+ T cells from healthy donors and treated the cells ex vivo with either of two shRNAs specific for TLR7 (clone 3 or 4), to silence TLR7, or with nontargeting shRNA, as a control (Fig. 6). After 2 d, we infected the shRNA-treated CD4+ T cells with concentrations of HIV-1NL-D within the physiological range34 (multiplicity of infection, 0.001) and measured the frequency of infected cells as well as their ability to produce proinflammatory cytokines every 48 h for a total of 11 d. Although CD4+ T cells transduced with nontargeting shRNA were infected with HIV-1NL-D to an extent similar to that of untransduced cells, cell populations transduced with TLR7-specific shRNA had a much lower frequency of HIV-1NL-D-infected CD4+ T cells at all time points examined (Fig. 6a,b). This lower frequency of HIV-1+ cells was not due to a specific increase in the death of cells in which TLR7 was silenced, as there was no difference between cell populations transduced with TLR7-specific shRNA and control cell populations transduced with nontargeting shRNA in terms of the frequency of early or late apoptotic CD4+ T cells after infection (Supplementary Fig. 8). We further confirmed the role of TLR7 in decreasing the HIV-1NL--D infection rate by blockade of TLR7 on CD4+ T cells from healthy donors with various doses of IRS 661 before in vitro infection (Fig. 6c,d). Moreover, the secretion of cytokines by HIV-1NL-D-infected cells was significantly different for cells transduced with nontargeting shRNA and their counterparts transduced with TLR7-specific shRNA (Fig. 6e,f). Although HIV-1NL-D-infected cells in cultures of CD4+ T cells transduced with nontargeting shRNA produced less IL-2 and IFN-γ protein than did mock-infected CD4+ T cells, the low frequency of HIV-1NL-D-infected CD4+ T cells transduced with TLR7-specific shRNA secreted significantly more IL-2 and IFN-γ than did their counterparts transduced with nontargeting shRNA (Fig. 6e,f), which suggested that the anergic phenotype did not develop after infection in cells transduced with TLR7-specific shRNA. To confirm that observation, we assessed the expression of anergy-related genes in CD4+ T cells obtained from healthy donors, infected with HIV-1NL-D in vitro in the presence of IRS 661 (to inhibit TLR7) or a control sequence and assessed at day 7 after infection. Sorted HIV-1NL-D+ CD4+ T cells showed higher expression of eight of the twelve anergy-related genes examined than did HIV-1NL-D- CD4+ T cells, while HIV-1+ CD4+ T cells sorted from IRS 661-treated cultures did not upregulate any of these genes at the time point analyzed (Fig. 6g). The expression of other genes encoding molecules that have been functionally linked to anergy, such as ITCH, DGKA, CBLB and SIRT1, was also upregulated in HIV-1NL-D+ CD4+ T cells but not in IRS 661-treated HIV-1NL-D+ T cells (data not shown). These data suggested that an interaction of HIV-1NL-D with TLR7 was responsible, at least in part, for the anergic phenotype observed in HIV-1NL-D-infected CD4+ T cells.
We investigated the role of the intracellular calcium- and NFATc2-activated gene-expression signaling events we observed by engagement of TLR7 with imiquimod, after infection with HIV-1NL-D in vitro. We hypothesized that blockade of intracellular calcium and silencing of NFATC2 would lead to a decrease in the frequency of HIV-1+ T cells even in the presence of a functional TLR7. To induce calcium blockade, we preincubated CD4+ T cells with the chelation agent Quin-2 AM36 before infection with HIV-1NL-D. Calcium chelation significantly decreased the frequency of viable cells, even at the lowest concentration of the chelating agent (Fig. 6h), perhaps due to the essential role of calcium in many cellular processes. Nevertheless, there was a lower frequency of HIV-1NL-D+ T cells among the remaining viable CD4+ T cells in cultures treated with Quin-2 AM than among CD4+ T cells treated with vehicle alone (Fig. 6h), in agreement with published investigations suggesting a role for calcium in the HIV-1 life cycle37. We then silenced NFATC2 with either of two different shRNAs or blocked NFATc2 with VIVIT peptide, which interferes with the calcineurin-NFATc2 interaction and inhibits dephosphorylation of NFAT38. In both cases, the absence of functional NFATc2 led to a significantly lower frequency of HIV-1NL-D-infected T cells than among CD4+ T cells left untreated or treated with vehicle alone (Fig. 6i,j), which indicated a role for NFATc2 in HIV-1 infection, as previously suggested39. These data suggested that calcium-dependent activation of NFATc2 upon triggering of TLR7 was involved in HIV-1NL-D infection in vitro.
Calcium-induced anergy favors HIV-1 replication
Given the decreased infection rate of cells in which TLR7 was silenced and the non-anergic phenotype of infected CD4+ T cells in which TLR7 was silenced, we hypothesized that the anergic state induced by stimulation via TLR7 during infection with HIV-1 would be a necessary step for HIV-1 to replicate in the host. In the absence of signaling via TLR7, the virus would not be able to render the infecting cell anergic and long-term infection would not occur. To test this hypothesis, we obtained CD4+ T cells from healthy donors, induced anergy in the cells via the TLR7 pathway with various doses of imiquimod and subsequently infected them with HIV-1NL-D and analyzed them 7 d later. Of note, the frequency of HIV-1NL-D-infected CD4+ T cells directly correlated with the concentration of imiquimod used and the increase in intracellular calcium (Fig. 7a,b), and thus with the degree of anergy in the culture, which suggested that calcium-induced anergy favored infection with HIV-1. Moreover, the induction of anergy via other well-established in vitro methods, such as treatment with ionomycin (Fig. 7c,d) or stimulation with anti-CD3 without costimulatory signals (Fig. 7e,f), before infection with HIV-1NL-D also increased the frequency of HIV-1NL-D-infected cells. These data supported the hypothesis that HIV-1-induced anergy via ligation of TLR7 and an increase in intracellular calcium concentration were prerequisites for productive infection with HIV-1.
To further investigate whether inhibiting TLR7-induced anergy would affect the frequency of HIV-1-infected cells, we added either the calcineurin inhibitor cyclosporine A or concentrations of IL-2 that have been shown to reverse anergy in several in vitro settings17 before in vitro infection of cells with HIV-1NL-D. Although the addition of IL-2 6 h before infection with HIV-1NL-D did not affect the frequency of HIV-1NL-D-infected cells (data not shown), perhaps due to response kinetics, the broad spectrum of IL-2 functions or the inability of IL-2 to 'rescue' CD4+ T cells from TLR7-induced anergy, blocking anergy with cyclosporine A significantly decreased the frequency of HIV-1NL-D+ T cells (Fig. 7g). This further supported the hypothesis that HIV-1-induced anergy was necessary for productive infection with HIV-1.
Inhibition of TLR7 diminishes HIV-1 in T cells ex vivo
We next sought to directly investigate the role of TLR7 in CD4+ T cells from HIV-1-infected patients. Specifically, on the basis of the in vitro model system with HIV-1NL-D, we hypothesized that inhibition of the TLR7 pathway in CD4+ T cells from HIV-1-infected patients would decrease the infection rate. We isolated CD4+ T cells from HIV-1 infected patients (Supplementary Table 1) and stimulated the cells in the presence of IRS 661 or transduced them with either of two TLR7-specific shRNAs, then collected supernatants every 3 d for a total of 14 d to measure virus concentration by ELISA of the HIV-1 core antigen p24. The inhibition of TLR7 by IRS 661 substantially decreased the concentration of p24 in culture (Fig. 8a,b), and we obtained similar results for cells transduced with TLR7-specific shRNA (Fig. 8c,d). We assayed CD4+ T cells from healthy donors in parallel as a negative control but found no detectable p24 at any time point (data not shown). Furthermore, we measured the load of proviral integrated DNA at day 7 after stimulation with anti-CD3 and anti-CD28 in the presence of IRS 661 or a control sequence and after transduction with TLR7-specific or nontargeting shRNA to monitor the cellular viral reservoir. Inhibition of TLR7 by either IRS 661 (Fig. 8e) or knockdown of TLR7 (Fig. 8f) resulted in a significantly lower proviral DNA load than that in control cells. Together these results showed that inhibition of TLR7 signaling was able to diminish the HIV-1 load in infected CD4+ T cells derived from infected patients.
Discussion
Microbial pattern-recognition receptors are critical for the early sensing of diverse bacterial and viral infections for activation of the innate immune system. Here we have described a previously unknown function of these pattern-recognition receptors in shutting off the immune responses of human CD4+ T cells. Engagement of TLR7 by its ligands in CD4+ T cells prevented entry into the cell cycle and the secretion of proinflammatory cytokines after stimulation. Mechanistically, stimulation of TLR7 increased intracellular calcium concentrations, which led to dephosphorylation of NFATc2 and its translocation to the cell nucleus; this activated an anergic gene-expression program. Furthermore, TLR7 signaling interfered with costimulatory signals, as activation of Jnk and Jun was inhibited in the presence of TLR7 ligands and full stimulation through the TCR and costimulation via CD28. Our results have potential clinical implications, as silencing of TLR7 inhibited the frequency of cells infected in vitro with HIV-1NL-D, and those infected cells did not display an anergic phenotype, which suggested that the calcium-induced TLR7-dependent anergic state of HIV-1NL-D-infected cells might have a role in HIV-1 persistence.
TLR signaling has been studied predominantly in APCs, in which engagement of the ligand induces the secretion of proinflammatory cytokines and upregulation of activation molecules12, 13, 14. Although TLR signaling in CD4+ T cells has not been studied in depth, the few reports published have demonstrated a positive costimulatory role for TLR signaling7, 8, 40. Our results have demonstrated a previously undescribed TLR signaling pathway with an inhibitory effect on T cell proliferation and cytokine secretion. The differences between the phenotypes of monocytes and those of CD4+ T cells upon TLR7 stimulation could be due to the intrinsic differences in activation requirements for each cell population. Although CD4+ T cells need engagement of the TCR and a costimulatory signal to enter cell cycle, CD14+ cells require one signal. Thus, engagement of TLR7 by its ligand is sufficient to induce activation and secretion of proinflammatory cytokines in monocytes, as expected for a cell of the innate immune system. In contrast, stimulation of TLR7 in CD4+ T cells leads to a significant increase in intracellular calcium that in the absence of a costimulatory signal triggers the TLR7-driven anergic program17. In this context, our results also showed a decrease in CD4+ T cell proliferation upon stimulation of TLR9, with no effect on IFN-γ secretion and a trend toward increased IL-17 secretion, although this result was not significant. Furthermore, there was no increase in the intracellular calcium concentration when CD4+ T cells were stimulated with CpG B, which suggested that the mechanism by which CpG B modifies CD4+ T cell functionality is not common to ligation of TLR7.
We note that all the ligands we used here were pharmacological agonists of TLR7. We have not been able to find any immunostimulatory RNA specific for human TLR7, which would be a more physiological ligand, as these immunostimulatory RNA sequences are specific for mouse TLR7 but recognize human TLR8 instead. We have overcome this issue through the use of HIV-1 as a potential 'natural' ligand of TLR7. The physiological relevance of our data is suggested by the effect of TLR7 signaling on HIV-1-infected cells. Several reports have highlighted the role of innate sensors in infection with HIV-1, focusing on the function of these molecules in CD4+ T cells that had not been productively infected41, 42. Our results add a new layer of complexity to the understanding of the HIV viral life cycle, which needs further investigation, and are in contrast with published data describing the cytoplasmic sensors of viral DNA that trigger cell death by pyroptosis of unproductively infected CD4+ T cells41, 42. Moreover, the observations reported here suggest involvement of TLR7 both in the induction of the anergic phenotype observed in HIV-1-infected cells and in increases in the degree of infection of CD4+ T cells, which suggests that TLR7-induced upregulation of calcium is involved in the viral life cycle. In this context, TLR8-dependent activation of NF-κB has been shown to be critical for HIV-1 replication in dendritic cells43. Although we did not investigate the role of TLR8 in the infection of CD4+ T cells by HIV-1, it is possible that contributions of both TLR7-driven activation of NF-κB and TLR7-induced anergy have a role in the increased viral load. The observation that anergy favored HIV-1 infection is somewhat paradoxical given existing literature showing that HIV-1 infection is favored in activated CD4+ T cells11. We speculate that HIV-1 might trigger TLR7 to induce an increase in intracellular calcium concentrations, which has been suggested to be important for the viral life cycle37. Whether the induction of anergy in host cells by HIV-1 infection via TLR7 is a bystander consequence of the increase in intracellular calcium driven by TLR7, and whether this status is beneficial for the host or not, are currently unknown. However, these observations open up a new field of investigation related to the mechanism, kinetics and consequences of the interaction of HIV-1 with the host cell through TLR7.
Although infections are generally regarded as illnesses, mammals are colonized with bacteria and viruses. The 'prime' example of this is bacteria adopted for digestion, although humans are also colonized with common DNA and RNA viruses, including endogenous retroviruses. It has been suggested that these endogenous retroviruses provide an adoptive selective advantage in generating genetic diversity44. On the basis of our data here, we speculate that TLR7 might have been co-opted in human CD4+ T cells to co-evolve so that the cells do not enter into the cell cycle in response to endogenous retroviruses, with potential consequences such as leukemia (HTLV-1) and autoimmune diseases. There are examples in the literature showing that certain endogenous retrovirus sequences are upregulated in human autoimmune diseases45. Although it will be of interest in future investigations to elucidate precisely when TLR7 is engaged in the life cycle of HIV-1 during infection, these data demonstrate a mechanism by which HIV-1 may avoid elimination by co-opting NFAT-dependent TLR7-induced T cell anergy.
In summary, we have demonstrated a previously unknown role for TLR7 in CD4+ T cell function that is in direct opposition to its role in cells of the innate immune system. Moreover, TLR7 ligands may be used as a means of inducing 'tolerance' on CD4+ T cells in human autoimmune diseases. Finally, our data have demonstrated a novel function for microbial pattern-recognition receptors in the inhibition of immune responses.
|
|
| |
| |
|
|
|