| |
Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial
|
| |
| |
Download the PDF here
Download the PDF here
RG-101 IN COMBINATION WITH 4 WEEKS OF ORAL DIRECT ACTING ANTIVIRAL THERAPY ACHIEVES HIGH VIROLOGIC RESPONSE RATES IN TREATMENT NAïVE GENOTYPE 1 AND 4 CHRONIC HEPATITIS C PATIENTS: INTERIM RESULTS FROM A RANDOMISED, MULTI-CENTER, PHASE 2 STUDY.......
http://www.natap.org/2016/EASL/EASL_14.htm
Regulus Reports Clinical Hold of RG-101.....
http://www.natap.org/2016/HCV/062716_05.htm
AASLD: Towards a Single Treatment Cure for HCV: Reformulation of the Pangenotypic NS5B NNI GSK2878175 as a Long Acting Parenteral (LAP) - (11/21/16)
AASLD: Regulus Expands Clinical Trial Collaboration with GSK ....."potential single-visit cure" - (11/29/16)
GSK Combination Study on Track; Data by Year-End 2016. In March 2016, Regulus plans to initiate a multi-center, open-label Phase II study evaluating the combination of a single subcutaneous injection of 4 mg/kg of RG-101 and daily oral administrations of 20 mg of GSK2878175, an investigational non-nucleoside NS5B polymerase inhibitor, for up to 12 weeks in treatment-naïve patients chronically infected with HCV genotypes 1 and 3. Concurrently, GSK will work on developing a "LAP" formulation of GSK2878175 as a single intra-muscular injection, providing the potential for a single-visit therapeutic treatment for HCV that could improve patient compliance through reduced dosing intervals and potentially extend opportunities for HCV therapeutic intervention. This LAP formulation of GSK2878175 may be used in additional clinical trials together with RG-101 following completion of the planned Phase II study, although any additional studies are not covered by the current collaboration agreement. Regulus expects to report safety and efficacy data from the GSK Phase II study before the end of 2016.
AASLD: A Single Subcutaneous Dose of 2 mg/kg or 4 mg/kg of RG-101, a Galnac-Conjugated Oligonucleotide with Antagonist Activity against miR-122, Results in Significant Viral Load Reductions in Chronic Hepatitis C Patients - (11/29/16)
"In conclusion, RG-101 administered in 2 mg/kg or 4 mg/kg in a single subcutaneous injection to patients with chronic HCV infection genotype 1, 3, and 4 appeared safe, provided dose-dependent plasma exposure of the drug, and resulted in significant viral load reduction in all treated patients within 4 weeks, and sustained virological response in three patients at week 76 of follow-up. Phase 2 studies are underway (eg, EudraCT 2015-001535-21) to establish the efficacy of a combination of RG-101 with direct-acting antivirals to potentially shorten treatment duration......At week 4, the median viral load reduction from baseline was 4⋅42 (IQR 3⋅23-5⋅00) and 5⋅07 (4⋅19-5⋅35) log10IU/mL in patients dosed with 2 mg/kg RG-101 or 4 mg/kg RG-101. Three patients had undetectable HCV RNA levels 76 weeks after a single dose of RG-101......One patient had transient increases in alkaline phosphatase and γ-glutamyl transferase levels, accompanied with increases in alanine aminotransferase, aspartate aminotransferase, and direct bilirubin levels.......miR-122 has a tumour suppressive role and low miR-122 levels have been related to the development of hepatocellular carcinoma.15,26,27,28Although the exact long-term risk of hepatocellular carcinoma development in patients with chronic hepatitis C after short-term miR-122 inhibition is unknown, a retrospective follow-up study of patients dosed with miravirsen showed no safety issues.29An extension study of the current study is ongoing (EudraCT 2016-002069-77) with the intent to follow up patients treated with RG-101 for up to 3 years after dosing to assess long-term safety."
----------------------
Safety, tolerability, and antiviral effect of RG-101 in patients with chronic hepatitis C: a phase 1B, double-blind, randomised controlled trial
Lancet Jan 10 2016
Meike H van der Ree, J Marleen de Vree, Femke Stelma, Sophie Willemse, Marc van der Valk, Svend Rietdijk, Richard Molenkamp, Janke Schinkel,
Ad C van Nuenen, Ulrich Beuers, Salah Hadi, Marten Harbers, Eva van der Veer, Kai Liu, John Grundy, Amy K Patick, Adam Pavlicek,
Jacqueline Blem, Michael Huang, Paul Grint, Steven Neben, Neil W Gibson, Neeltje A Kootstra, Hendrik W Reesink
Summary
Background
miR-122 is an important host factor for hepatitis C virus (HCV) replication. The aim of this study was to assess the safety and tolerability, pharmacokinetics, and antiviral effect of a single dose of RG-101, a hepatocyte targeted N-acetylgalactosamine conjugated oligonucleotide that antagonises miR-122, in patients with chronic HCV infection with various genotypes.
Methods
In this randomised, double-blind, placebo-controlled, multicentre, phase 1B study, patients were randomly assigned to RG-101 or placebo (7:1). We enrolled men and postmenopausal or hysterectomised women (aged 18-65 years) with chronic HCV genotype 1, 3, or 4 infection diagnosed at least 24 weeks before screening who were either treatment naive to or relapsed after interferon-α based therapy. Patients with co-infection (hepatitis B virus or HIV infection), evidence of decompensated liver disease, or a history of hepatocellular carcinoma were excluded. Randomisation was done by an independent, unblinded, statistician using the SAS procedure Proc Plan. The first cohort received one subcutaneous injection of 2 mg/kg RG-101 or placebo; the second cohort received one subcutaneous injection of 4 mg/kg or placebo. Patients were followed up for 8 weeks (all patients) and up to 76 weeks (patients with no viral rebound and excluding those who were randomised to the placebo group) after randomisation. The primary objective was safety and tolerability of RG-101. This trial was registered with EudraCT, number 2013-002978-49.
Findings
Between June 4, 2014, and Oct 27, 2014, we enrolled 32 patients with chronic HCV genotype 1 (n=16), 3 (n=10), or 4 (n=6) infections. In the first cohort, 14 patients were randomly assigned to receive 2 mg/kg RG-101 and two patients were randomly assigned to receive placebo, and in the second cohort, 14 patients were randomly assigned to receive 4 mg/kg RG-101 and two patients were randomly assigned to receive placebo. Overall, 26 of the 28 patients dosed with RG-101 reported at least one treatment-related adverse event. At week 4, the median viral load reduction from baseline was 4⋅42 (IQR 3⋅23-5⋅00) and 5⋅07 (4⋅19-5⋅35) log10 IU/mL in patients dosed with 2 mg/kg RG-101 or 4 mg/kg RG-101. Three patients had undetectable HCV RNA levels 76 weeks after a single dose of RG-101. Viral rebound at or before week 12 was associated with the appearance of resistance associated substitutions in miR-122 binding regions in the 5' UTR of the HCV genome.
Interpretation
This study showed that one administration of 2 mg/kg or 4 mg/kg RG-101, a hepatocyte targeted N-acetylgalactosamine conjugated anti-miR-122 oligonucleotide, was well tolerated and resulted in substantial viral load reduction in all treated patients within 4 weeks, and sustained virological response in three patients for 76 weeks.
Discussion
In this study, one dose of 2 mg/kg or 4 mg/kg of RG-101 was well tolerated and no serious adverse events were noted in patients with chronic hepatitis C. Treatment with RG-101 led to a substantial decrease in HCV RNA levels in all patients with chronic hepatitis C infected with HCV genotype 1, 3, and 4. Median viral load reductions at week 4 were 4⋅42 log10 IU/mL for 2 mg/kg treatment and 5⋅07 log10 IU/mL for 4 mg/kg treatment and HCV RNA levels were undetectable in three patients at week 76 of follow-up after one dose of RG-101.
Other host-targeting agents that have been studied previously in HCV infection include alisporivir, a cyclophilin A inhibitor, and miravirsen. Clinical development of alisporivir was stopped because of concerns about development of pancreatitis. Previous studies with miravirsen showed potential for therapeutic efficacy of a miR-122 inhibitor in patients who were treatment naive with HCV genotype 1. A transient viral load reduction of more than 2 log10 IU/mL occurred in 13 of 27 patients dosed with 3 mg/kg, 5 mg/kg, or 7 mg/kg miravirsen.9 RG-101 is the first in-human tested anti-miR oligonucleotide that is targeted for delivery to hepatocytes with N-acetylgalactosamine, a high-affinity ligand for the asialoglycoprotein receptor that is abundantly expressed on hepatocytes. N-acetylgalactosamine conjugation of an oligonucleotide results in enhanced targeting to hepatocytes and lower exposure in non-hepatic organs.25
In this study, there were no clinically significant findings with respect to the clinical safety laboratory tests described in the Methods. On the basis of clinical and non-clinical data with other antisense oligonucleotides, special attention was given to potential risks of complement activation, increased liver enzymes, activated partial thromboplastin time, C-reactive protein, and renal safety. As expected from earlier studies with miR-122 inhibitors,9, 12, 14, 20 RG-101 administration resulted in a modest and prolonged increase in serum alkaline phosphatase levels and a prolonged decrease in total plasma cholesterol levels in patients with chronic hepatitis C. The alkaline phosphatase gene (ALPL) contains multiple miR-122 seed matches in the 3' UTR and is considered as a direct target of miR-122. Anti-miR-122 dosing resulted in hepatic ALPL de-repression in animal studies, and elevated serum alkaline phosphatase levels were noted in miR-122 knockout mice.26, 27 One patient had transient increases in alkaline phosphatase and γ-glutamyl transferase levels, accompanied with increases in alanine aminotransferase, aspartate aminotransferase, and direct bilirubin levels. A possible relationship with alcohol consumption could not be excluded and future research might be needed to assess the effects of alcohol on treatment with RG-101. miR-122 has a tumour suppressive role and low miR-122 levels have been related to the development of hepatocellular carcinoma.15, 26, 27, 28 Although the exact long-term risk of hepatocellular carcinoma development in patients with chronic hepatitis C after short-term miR-122 inhibition is unknown, a retrospective follow-up study of patients dosed with miravirsen showed no safety issues.29 An extension study of the current study is ongoing (EudraCT 2016-002069-77) with the intent to follow up patients treated with RG-101 for up to 3 years after dosing to assess long-term safety.
Animal studies have shown that RG-101 is rapidly absorbed into the systemic circulation and cleared from the plasma within 24 h to various tissues that include the liver.30 Once taken up by hepatocytes, RG-101 is efficiently metabolised to its main active metabolite (RG1649), which has a tissue half-life in animals of about 14 days. The findings in patients with chronic hepatitis C were consistent with findings in animal studies showing that RG-101 is stable in plasma and is cleared from plasma within 24 h, which most probably reflects extensive tissue uptake (including liver), rather than extensive metabolism or urinary elimination. The tissue half-life of RG-101 was not assessed in humans, but might be similar to that in animals given the prolonged pharmacodynamic effects (increased alkaline phosphatase levels and decreased cholesterol levels) and prolonged antiviral effect. In this proof-of-concept study, there were three patients with undetectable HCV RNA levels at 76 weeks after administration of one dose of RG-101. According to HCV guidelines, in which a sustained virological response is defined as undetectable HCV RNA levels at 12 weeks or 24 weeks after completion of therapy,3 these patients could be considered HCV cured. However, these results should be interpreted with caution considering the distinct mechanism of action and long tissue half-life of RG-101 compared with HCV direct-acting antivirals. Despite the finding that these three patients had sustained virological responses 76 weeks after a dose of RG-101, RG-101 should probably not be used as monotherapy but should be combined with direct-acting antivirals. An in-vitro study31 showed that combination treatment with anti-miR-122 and direct-acting antiviral had additive or synergistic antiviral effects.
Monotherapy with HCV direct-acting antivirals rapidly selects for resistant viruses, whereas host-targeting agents have a relatively high barrier to resistance compared with most direct-acting antivirals. In this study, we showed that early virological rebound (<12 weeks) after RG-101 dosing was associated with the emergence of a single RAS (C3U) in patients infected with HCV genotype 1, as has been noted previously in patients dosed with miravirsen.30 Additionally, we showed emergence of four nucleotide changes including two RASs (C2G and C3U) and two polymorphisms (G1A and U4A) in the 5' UTR in patients with HCV genotype 3 or 4 with viral rebound after RG-101 dosing. Because the RASs appear in functional miR-122 binding regions,23 the emergence of these viral variants might alter the dependency of the virus on miR-122, however, this premise needs to be elucidated in future studies. A previous study32 showed that the C3U RAS in the 5' UTR of HCV confers reduced susceptibility to miR-122 antagonists but has no effect on the susceptibility to various direct-acting antivirals, thus providing support for testing regimens combining RG-101 and one or more direct-acting antivirals. In patients with a virological rebound more than 12 weeks after RG-101 dosing, we observed no emergence of RASs in miR-122 binding regions, and viral rebound could probably result from low (subtherapeutic) RG-101 drug concentrations in the liver.
A limitation of this study is that women of childbearing potential were excluded from participation, and therefore the study results might not be representative for this group of patients. Another limitation is that patients and investigators were unblinded for treatment allocation after week 8 of the study. This process might have influenced the adverse event reporting in the extended follow-up study.
The N-acetylgalactosamine linking to other oligonucleotides may have great potential for other liver diseases, such as chronic hepatitis B virus infection.33 The combination of a highly potent HCV direct-acting antiviral with RG-101 could potentially shorten HCV treatment duration to 4-6 weeks or less, which is of particular interest in view of the high costs of HCV direct-acting antivirals and potential patient non-adherence. An alternative application of miR-122 inhibitors, given the distinct mechanism of action, could be found in difficult to treat patients who previously did not respond to direct-acting antiviral therapy.
In conclusion, RG-101 administered in 2 mg/kg or 4 mg/kg in a single subcutaneous injection to patients with chronic HCV infection genotype 1, 3, and 4 appeared safe, provided dose-dependent plasma exposure of the drug, and resulted in significant viral load reduction in all treated patients within 4 weeks, and sustained virological response in three patients at week 76 of follow-up. Phase 2 studies are underway (eg, EudraCT 2015-001535-21) to establish the efficacy of a combination of RG-101 with direct-acting antivirals to potentially shorten treatment duration.
Funding
Regulus Therapeutics, Inc.
Introduction
Hepatitis C virus (HCV) is an enveloped, single-stranded RNA virus of the Flaviviridae family.1 Most people infected with HCV develop a chronic hepatitis C infection. Patients with this infection are at risk of developing complications such as liver cirrhosis and hepatocellular carcinoma.2 The goal of chronic hepatitis C treatment is to achieve a sustained virological response, defined as undetectable HCV RNA in blood 12-24 weeks after completing a course of antiviral treatment,3 which is associated with a reduced occurrence of liver-related complications and a prolonged survival.4, 5 Most new anti-HCV drugs are direct-acting antivirals that target specific viral proteins, such as NS3A protease, NS5A protein, and NS5B polymerase.
Present treatment regimens with a combination of direct-acting antivirals for 12 to 24 weeks result in high, sustained, virological response rates in most patients with chronic hepatitis C. Although treatment with a combination of direct-acting antivirals for 8 weeks in a selected group of patients with chronic hepatitis C (ie, HCV genotype 1, treatment naive, and non-cirrhotic) results in high, sustained, virological response rates, attempts to shorten treatment duration to less than 8 weeks with a triple or quadruple direct-acting antiviral regimen has shown high variability of sustained virological response rates.6, 7 An alternative treatment strategy is interference with host cell components that are essential for replication of HCV, such as cyclophilin A or microRNA (miRNA)-122.8, 9 Host-targeting agents have broad pan-genotypic antiviral activity,10 and have a relatively high genetic barrier to resistance compared with direct-acting antivirals. Furthermore, host-targeting agents might act in a synergistic manner with direct-acting antivirals resulting in shortened direct-acting antiviral therapy, including in those patients who are difficult to treat with present direct-acting antivirals.11
Research in context
Evidence before this study
This study was designed in 2013, before any all-oral regimens for hepatitis C virus (HCV) infection had been approved. We did PubMed searches for articles using the search terms "hepatitis C virus", "miRNA-122", and "antagomir". There were no language restrictions for this search. One previous randomised controlled study had shown therapeutic effect by targeting the host-factor miR-122 in patients with chronic HCV genotype 1 infection.
Added value of this study
This study assessed the safety, tolerability, pharmacokinetics, and antiviral effect of RG-101, a hepatocyte targeted oligonucleotide that antagonises miR-122, in patients with chronic HCV genotype 1, 3, and 4 infection. The results show that a dose of 2 mg/kg or 4 mg/kg of RG-101 was well tolerated and suggests an antiviral effect in HCV genotype 1, 3, and 4 infection. There were three patients with undetectable HCV RNA level at 76 weeks after a dose of RG-101. This study is the first, to our knowledge, to suggest that an anti-miR oligonucleotide linked to an N-acetylgalactosamine structure might have clinical efficacy.
Implications of all the available evidence
RG-101 appears to be more potent than a non-conjugated oligonucleotide against microRNA-122. The combination of a highly potent direct-acting antiviral with a host-targeting agent such as RG-101 could potentially shorten HCV treatment duration. In this phase 1B trial, RG-101 seems to be safe and well tolerated. Overall safety will be further evaluated in larger, late phase randomised controlled clinical trials.
miR-122 is an abundant, liver-specific miRNA that modulates the expression of genes involved in cholesterol and triglyceride metabolism12, 13, 14 and in tumorigenesis.15, 16, 17, 18 Additionally, miR-122 is a host factor that is essential for HCV replication. miR-122 binds to the 5' untranslated region (5' UTR) of the HCV genome and thereby promotes HCV RNA stability and accumulation,19, 20 protects HCV RNA against degradation by cellular exoribonucleases 1 and 2 (Xrn1 and Xrn2),21, 22, 23 and induces an increase in rate of viral RNA synthesis, or a combination of these factors.24 Previously, repeated dosing with miravirsen, a locked nucleic acid DNA phosphorothioate oligonucleotide inhibitor of miR-122, resulted in a dose-dependent and prolonged decrease in HCV RNA levels in patients with chronic hepatitis C with HCV genotype 1 infection.9 RG-101 is a mixed chemistry phosphorothioate oligonucleotide inhibitor of miR-122 that is linked to a multivalent N-acetylgalactosamine carbohydrate structure designed to enhance uptake of the oligonucleotide by hepatocytes through binding to the asialoglycoprotein receptor on hepatocytes.25 Conjugation to the N-acetylgalactosamine structure increases the potency of RG-101 by up to 10-20 times compared with the non-conjugated oligonucleotide. The aim of this proof-of-concept study was to assess the safety and tolerability, pharmacokinetics, and antiviral effect of one dose of an N-acetylgalactosamine conjugated oligonucleotide against miR-122 (RG-101) in patients with chronic HCV genotype 1, 3, and 4 infection.
Methods
Study design
In this randomised, double-blind, placebo-controlled, multicentre, phase 1B trial, we enrolled 32 patients aged 18-65 years with chronic hepatitis C at two hospitals in the Netherlands (Academic Medical Center, Amsterdam and University Medical Center, Groningen, the Netherlands). Liver fibrosis was assessed by liver elastography (transient elastography, known by the brand name FibroScan).
On study day 1, the first cohort received one subcutaneous injection of 2 mg/kg (volume injection range 0⋅9-1⋅5 mL) RG-101 or placebo, and the second cohort received one subcutaneous injection of 4 mg/kg (volume injection range 1⋅4-3⋅9 mL, or placebo). In the main study, both dosing cohorts were followed up for 8 weeks after randomisation (appendix). At the end of the main study, an independent physician determined which patients qualified to be enrolled in an extended follow-up study in which patients were followed up for a period of up to 76 weeks after dosing. At week 8, all patients treated with placebo or with virological rebound confirmed at retest were not included in the extended follow-up study. Virological rebound was defined as more than 1 log10 increase in HCV RNA level from nadir (defined as the lowest HCV RNA level after baseline). During the extended follow-up study, patients with virological rebound were excluded from further follow-up.
The study was approved by the regulatory authority and the independent ethics committee at each participating site, and was done in compliance with the Declaration of Helsinki, Good Clinical Practice guidelines, and local regulatory requirements. An independent data and safety monitoring committee reviewed the safety of the patients in the study.
Patients
Men and postmenopausal or hysterectomised women (aged 18-65 years) with chronic HCV genotype 1, 3, or 4 infection diagnosed at least 24 weeks before screening were enrolled. Eligible patients were treatment naive to, or relapsed after, interferon-α based therapy. Other eligibility criteria were those with a platelet count of more than 100 x 109/L, a total white blood cell count of more than 3⋅0 x 109/L, a haemoglobin concentration of more than 6⋅8 mmol/L for women and more than 7⋅4 mmol/L for men, an alanine aminotransferase concentration of less than five times the upper limit of normal (ULN), a total and direct bilirubin level within normal limits, a creatinine level within normal limits, and serum HCV RNA ≥75 000 IU/mL at screening. Patients with co-infection (hepatitis B virus or human immunodeficiency virus infection), evidence of decompensated liver disease, or a history of hepatocellular carcinoma were excluded (appendix). Before enrolment and before any study procedure, written informed consent was obtained from all patients.
Randomisation and masking
In each dose cohort (2 mg/kg or 4 mg/kg), patients were randomly assigned to receive either RG-101 or placebo (7:1; appendix). Allocation to treatment groups (placebo or active) was done randomly by an independent, unblinded, statistician, using the SAS procedure Proc Plan. Randomisation was done in blocks of eight patients, in a ratio of 1:7 (placebo:active). The randomisation list was kept by the unblinded pharmacist, and only provided to the study team after database hardlock and unblinding of the study at 8 weeks. Investigators and patients were blinded to treatment allocation and on-study HCV RNA results at least 8 weeks after RG-101 dosing.
Outcomes
The primary objective was to assess the safety and tolerability of one dose of subcutaneous RG-101 administered to patients with chronic HCV genotype 1, 3, and 4 infection. The secondary objective was to assess the pharmacokinetic profile and antiviral effect of RG-101 in patients with chronic hepatitis C.
Procedures
Safety was assessed in patients by physical examination, review of adverse events, laboratory testing of blood samples, and urinalysis (appendix). Calculated plasma and urine pharmacokinetic parameters included the maximum plasma concentration (Cmax), the time at which the maximum plasma concentration occurred (Tmax), the area under the plasma concentration curve up to the last measurable concentration (AUC0-t), and the percentage of the dose excreted in urine (as parent drug or major metabolite) during the first 24 h after receiving the treatment dose. Serum HCV RNA levels were measured with the Roche COBAS AmpliPrep/COBAS Taqman HCV (version 2.0) assay, with a reported lower limit of quantification of 15 IU/mL. Sustained virological response was defined as undetectable HCV RNA levels at 76 weeks after RG-101 treatment. HCV genotype was identified by sequence analysis of a fragment of the NS5B gene. Genotype of IFNL3 single nucleotide polymorphism rs12979860 was identified by in-house, developed, high-resolution melting curve analysis. Sequence analysis of the HCV 5' UTR of HCV RNA was done by 5' rapid amplification of complementary DNA ends (5' RACE System, version 2.0), followed by sequencing and was done at baseline for all patients and at time of virological rebound (>1 log10 increase in HCV RNA from nadir) and with HCV RNA of more than 20 000 IU/mL (appendix). In patients with HCV RNA levels below the detection limit for sequence analysis at time of rebound, a retest sample was used or an additional follow-up sample was collected.
Statistical analysis
All patients were analysed in the treatment group for which they were originally allocated after randomisation. For the analysis of safety and tolerability, and antiviral effect of RG-101 all patients were included. Patients dosed with placebo were excluded for the pharmacokinetic analysis. All statistics were descriptive, and calculated for each treatment group and included measurement of HCV RNA levels, clinical safety laboratory tests, and pharmacokinetics. All HCV RNA levels were analysed after log10 transformation. Samples with HCV RNA levels below the limit of quantification were set to an arbitrary HCV RNA level of 7⋅5 IU/mL and changes in HCV RNA levels as compared with baseline (day 1) were calculated. Statistics were done with SPSS (version 22.0) and GraphPad Prism Software (version 6.0). The trial was registered with EudraCT, number 2013-002978-49.
Role of the funding source
The study was designed and done by the funder (Regulus Therapeutics) in collaboration with PRA Health Sciences and the principal investigators. PRA Health Sciences and the principal investigators collected the data, monitored study conduct, and analysed the data. The corresponding author had full access to all data in the study and had final responsibility for the decision to submit for publication.
Results
Screening began on June 4, 2014, with the last patient enrolled on Oct 27, 2014. Of 41 patients screened, 32 were randomly assigned and received the study drug or placebo (figure 1). No protocol violations were reported. Overall, most patients were male (24 [75%] of 32), white (29 [91%] of 32), and treatment naive to interferon-α based therapy (24 [75%] of 32; table 1). Most patients were infected with HCV genotype 1 (n=16), followed by genotype 3 (n=10), and genotype 4 (n=6). Almost half of the patients (n=15) had fibrosis stage F0-F1 (table 1).
No serious adverse events, including deaths, occurred during the main study or the extended follow-up study that were considered to be related to RG-101 administration. Overall, 26 (93%) of the 28 patients dosed with RG-101 reported at least one treatment-related adverse event (table 2). The most common adverse events were fatigue, emotional distress, insomnia, and local injection site reactions. The injection site reactions were mild and self-limiting. One patient, a 46-year-old man, who received a dose of 4 mg/kg RG-101 developed a severe intrahepatic cholestasis, with an alanine aminotransferase level of 219 U/L (ULN 68 U/L), aspartate aminotransferase level of 235 U/L (45 U/L), γ-glutamyl transferase level of 6557 U/L (59 U/L), alkaline phosphatase level of 481 U/L (129 U/L), total bilirubin level of 49 μmol/L (29 μmol/L), and direct bilirubin level of 38 μmol/L (7 μmol/L), most probably as a consequence of alcohol abuse; however, a role for RG-101 cannot be excluded here. This patient was followed up at the outpatient clinic and all laboratory sample abnormalities improved after alcohol intake was stopped.
At week 4, median serum alanine aminotransferase and median serum aspartate aminotransferase levels had normalised (table 3; appendix). An increase in median serum alkaline phosphatase levels was noted in patients in both dosing groups at week 8 (table 3, appendix). Of the 28 patients treated with RG-101, six patients in the 2 mg/kg group (up to 2⋅0 x ULN) and seven patients in the 4 mg/kg group (up to 3⋅7 x ULN) had one or more alkaline phosphatase values of more than the ULN (appendix). Median alkaline phosphatase values had not yet returned to baseline values at the time of follow-up (appendix). There was no significant change in median total serum bilirubin levels (appendix). There were no clinically significant findings with respect to clinical safety laboratory tests (including clinical chemistry, haematological tests, coagulation, C-reactive protein, and urinalysis), complement activation, renal safety, vital signs, electrocardiograph, or physical examination after dosing with RG-101 (data not shown). A decrease in total plasma cholesterol levels was noted with a median reduction of 1⋅4 mmol/L (IQR 0⋅6-1⋅9) in patients dosed with 2 mg/kg RG-101 and 0⋅9 mmol/L (0⋅7-1⋅3) in patients dosed with 4 mg/kg RG-101 at week 4 compared with baseline, whereas median total cholesterol in patients treated with placebo increased compared with baseline (0⋅5 mmol/L [0⋅1-0⋅8]; appendix). Concentrations of HDL, LDL, and triglycerides all decreased proportionally in patients dosed with RG-101 (data not shown). At week 4, there was no correlation between change in HCV RNA level and total cholesterol level in patients dosed with 2 mg/kg RG-101 and 4 mg/kg RG-101 (R 0⋅34, p=0⋅076, appendix).
RG-101 was rapidly absorbed after subcutaneous administration, with a median plasma Tmax of 4 h for patients dosed with either 2 mg/kg or 4 mg/kg (appendix). After peaking, RG-101 clearance from plasma resulted in very low plasma concentrations (<0⋅17 μg/mL) observed by 24 h after administration (figure 2). Geometric mean Cmax and AUC0-t values for RG-101 increased more than dose proportional when increasing the dose from 2 mg/kg to 4 mg/kg (appendix). During the 0-24 h after drug administration time period, only the parent drug (RG-101) was observed in plasma, and less than 3% of the administered dose was eliminated as parent drug or its main active metabolite (RG1649) in the urine (appendix). No readily apparent differences in the plasma exposure of RG-101 were noted between treated patients with HCV who achieved HCV RNA levels below the limit of quantification and those who did not (data not shown).
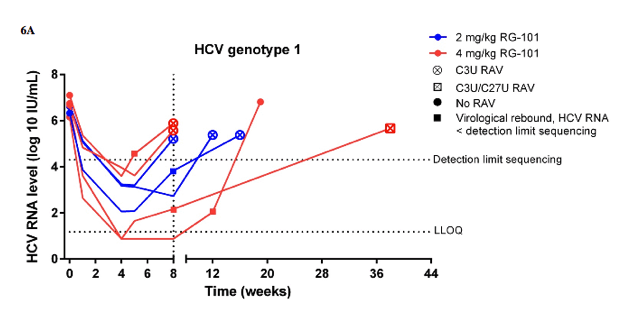
Baseline HCV RNA levels were similar between the RG-101 study groups (table 1). HCV RNA levels decreased rapidly in both dosing groups with a median decline of 2⋅63 log10 IU/mL (IQR 1⋅61-3⋅48) in patients dosed with 2 mg/kg RG-101 and 3⋅31 log10 IU/mL (2⋅57-4⋅01) in patients with 4 mg/kg RG-101 at week 1 compared with baseline (appendix). At week 4, the median reduction in HCV RNA level from baseline was 4⋅42 log10 IU/mL (IQR 3⋅23-5⋅00) in patients dosed with 2 mg/kg RG-101 and 5⋅07 log10 IU/mL (4⋅19-5⋅35) in patients dosed with 4 mg/kg RG-101 (figure 3A). Of the 14 patients treated with 2 mg/kg RG-101, four had a virological rebound at week 8 and the other ten were included in the extended follow-up study (figure 1, figure 3B). Of patients dosed with 4 mg/kg RG-101, two patients had a virological rebound at week 8 and the other 12 patients were included in the extended follow-up study (figure 1, figure 3C). Of those who were included in the extended follow-up study, 15 of 22 patients had an HCV RNA level below the limit of quantification at week 8 (HCV genotype 1 [n=6], genotype 3 [n=5], and genotype 4 [n=4]). In the extended follow-up study, 16 of 22 patients had a virological rebound; these occurred at week 12 (n=8), week 16 (n=2), week 20 (n=1), week 28 (n=2), week 36 (n=2), and week 52 (n=1). Three patients had a sustained virological response at week 76 after one dose of RG-101; one patient with HCV genotype 3 and IFNL3 genotype CC who was treated with 2 mg/kg RG-101 and two patients with HCV genotype 4 and IFNL3 genotype CC or CT both treated with 4 mg/kg RG-101 (figures 3B, 3C). The remaining three patients withdrew consent (n=2) or were lost to follow-up by week 76 (n=1), of whom one had undetectable HCV RNA level at week 12 (figure 1).
At baseline, none of the patients had a mutation in any of the miR-122 binding sites located in the 5' UTR of the HCV genome (S1, S2, or additional base-pair interactions) compared with the reference sequence (data not shown). In 14 of 22 RG-101 treated patients who had a viral rebound, matched samples (at baseline and time of viral rebound) with viral loads of more than 20 000 IU/mL were available. Among these 14 patients, six patients with HCV genotype 1 with viral rebound between weeks 5 and weeks 12 had a resistance associated substitution (RAS; figure 4, appendix). A single RAS, namely C3U, in the 5' UTR miR-122 binding region was noted in five of these six patients. One of the six patients had viral rebound at week 8 (HCV RNA <20 000 IU/mL) and only a week 38 sample was available for analysis. This week 38 sample contained the C3U RAS and also a mixed population of C and U at position 27 (appendix). Additionally, two patients (HCV genotype 3 and 4) had a viral rebound between weeks 8 and weeks 12, and had HCV variants containing four nucleotide changes, including two RASs (C2G and C3U) and two polymorphisms (G1A and U4A) in the 5' UTR miR-122 binding region (figure 4, appendix). The remaining six patients (HCV genotype 1 [n=1], 3 [n=4], and 4 [n=1]) had viral rebound between weeks 12 and 36 and had no RASs in the 5' UTR miR-122 binding regions (figure 4, appendix). An additional polymorphism (G38A) was identified in one patient (HCV genotype 4) with viral rebound at week 20. There was good agreement between population based and deep sequence analyses (data not shown).
|
|
| |
| |
|
|
|