| |
Glecaprevir and pibrentasvir for 12 weeks for hepatitis C virus genotype 1 infection and prior direct-acting antiviral treatment
|
| |
| |
Download the PDF here
EASL/2016: HIGH EFFICACY OF ABT-493 AND ABT-530 IN HCV GENOTYPE 1-INFECTED PATIENTS WHO HAVE FAILED DIRECT-ACTING ANTIVIRAL-CONTAINING REGIMENS: THE MAGELLAN-I STUDY - (04/15/16)
"In part 1 of the MAGELLAN-1 study, we evaluated the efficacy and safety of GLE + PIB for 12 weeks, with or without RBV, in patients with prior treatment failure of HCV regimens containing an NS5A inhibitor and/or NS3/4A PI with or without NS5B inhibitors. The impacts of baseline polymorphisms and RBV coadministration on SVR12 rates were also assessed......baseline NS5A polymorphisms were detected in 50% (3/6), 50% (11/22), and 55% (12/22) of patients in arms A, B, and C, respectively, at a detection threshold of 15%. Although a detection threshold of 15% is commonly accepted as clinically relevant, lower thresholds of detection give greater insight into amino acid changes that may emerge in the majority of the population after prior therapy with DAA-containing regimens. At a more sensitive 2% detection threshold, the most common polymorphisms in NS3 were at amino acid positions Q80 (n = 23), R155 (n = 4), D168 (n = 4), and I/V170 (n = 4); and those for NS5A were at positions Q30 (n = 14), Y93 (n = 8), L31 (n = 7), M28 (n = 6), Q54 (n = 6), and H/P58 (n = 5). Baseline amino acid polymorphisms detected at a threshold of 2% are shown in Supporting Table S2, and the prevalence of specific polymorphisms detected at both 2% and 15% is summarized in Supporting Table S3......Overall, in patients with baseline polymorphisms in NS3 only, NS5A only, or both NS3 and NS5A at a 15% detection threshold, SVR12 rates were 100% (14/14), 91% (10/11), and 93% (14/15), respectively; SVR12 was achieved by 100% (10/10) of patients with no baseline polymorphisms in NS3 or NS5A.....This confirms in vitro data that variants at the Y93 position are susceptible to PIB[22] and suggests GLE plus PIB is an effective treatment for those with a baseline polymorphism or treatment-emergent substitution at this position. Furthermore, 14 of 15 (93%) patients who failed a prior dual (NS3/4A PI plus NS5A inhibitor) or triple (NS3/4A PI plus NS5A inhibitor plus NS5B polymerase inhibitor) DAA regimen achieved SVR12â€
------------------------------
Abbvie 2-drug - ABT493+ABT530, waiting for FDA approval, around August expected
AASLD:ENDURANCE-1: A Phase 3 Evaluation of the Efficacy and Safety of 8- versus 12-week Treatment with Glecaprevir/Pibrentasvir (formerly ABT-493/ABT-530) in HCV Genotype 1 Infected Patients with or without HIV-1 Co-infection and without Cirrhosis - (11/15/16)
AASLD:ENDURANCE-2: Safety and Efficacy of Glecaprevir/Pibrentasvir in Hepatitis C Virus Genotype 2-infected Patients without Cirrhosis: a Randomized, Double-Blind, Placebo-Controlled Study - (11/14/16)
AASLD:EXPEDITION-4: EFFICACY AND SAFETY OF GLECAPREVIR/PIBRENTASVIR (ABT-493/ABT-530) IN PATIENTS WITH RENAL IMPAIRMENT AND CHRONIC HEPATITIS C VIRUS GENOTYPE 1 - 6 INFECTION - (11/15/16)
AASLD:SURVEYOR-II, PART 3: EFFICACY AND SAFETY OF GLECAPREVIR/PIBRENTASVIR (ABT-493/ABT-530) IN PATIENTS WITH HEPATITIS C VIRUS GENOTYPE 3 INFECTION WITH PRIOR TREATMENT EXPERIENCE AND/OR CIRRHOSIS - (11/14/16)
AASLD:SURVEYOR-II, Part 4: Glecaprevir/Pibrentasvir Demonstrates High SVR Rates in Patients With HCV Genotype 2, 4, 5, or 6 Infection Without Cirrhosis Following an 8-Week Treatment Duration - (11/16/16)
AASLD:ENDURANCE-4: Efficacy and Safety of Glecaprevir/Pibrentasvir (Formerly ABT-493/ABT-530) Treatment in Patients with Chronic HCV Genotype 4, 5, or 6 Infection - (11/14/16)
----------------------
Glecaprevir and pibrentasvir for 12 weeks for hepatitis C virus genotype 1 infection and prior direct-acting antiviral treatment
Hepatology Epub Jan 27, 2017 - Fred Poordad,1 Franco Felizarta,2 Armen Asatryan,3 Mark S. Sulkowski,4 Robert W. Reindollar,5 Charles S. Landis,6 Stuart C. Gordon,7 Steven L. Flamm,8 Michael W. Fried,9 David E. Bernstein,10 Chih-Wei Lin,3 Ran Liu,3 Sandra S. Lovell,3 Teresa I. Ng,3 Jens Kort,3 and Federico J. Mensa3
Abstract
Although direct-acting antiviral (DAA) therapies for chronic hepatitis C virus (HCV) infection have demonstrated high rates of sustained virologic response, virologic failure may still occur, potentially leading to the emergence of viral resistance, which can decrease the effectiveness of subsequent treatment. Treatment options for patients who failed previous DAA-containing regimens, particularly those with nonstructural protein 5A inhibitors, are limited and remain an area of unmet medical need. This phase 2, open-label study (MAGELLAN-1) evaluated the efficacy and safety of glecaprevir (GLE) + pibrentasvir (PIB) ± ribavirin (RBV) in HCV genotype 1–infected patients with prior virologic failure to HCV DAA-containing therapy. A total of 50 patients without cirrhosis were randomized to three arms: 200 mg GLE + 80 mg PIB (arm A), 300 mg GLE + 120 mg PIB with 800 mg once-daily RBV (arm B), or 300 mg GLE + 120 mg PIB without RBV (arm C). By intent-to-treat analysis, sustained virologic response at posttreatment week 12 was achieved in 100% (6/6, 95% confidence interval 61-100), 95% (21/22, 95% confidence interval 78-99), and 86% (19/22, 95% confidence interval 67-95) of patients in arms A, B, and C, respectively. Virologic failure occurred in no patients in arm A and in 1 patient each in arms B and C (two patients were lost to follow-up in arm C). The majority of adverse events were mild in severity; no serious adverse events related to study drug and no relevant laboratory abnormalities in alanine aminotransferase, total bilirubin, or hemoglobin were observed. Conclusion: The combination of GLE and PIB was highly efficacious and well tolerated in patients with HCV genotype 1 infection and prior failure of DAA-containing therapy; RBV coadministration did not improve efficacy.
Hepatitis C virus (HCV) genotype (GT) 1 is the most common among HCV GTs globally, accounting for approximately 46% of an estimated 185 million infections worldwide.[1, 2] Although the treatment landscape for HCV has rapidly evolved with highly effective and safe treatments for patients with GT1 infection,[3, 4] as direct-acting antiviral (DAA) agents are used extensively, the number of patients with virologic failure of DAA regimens continues to grow.[5] Virologic failure of DAAs often results from baseline resistance-associated polymorphisms or resistance-associated substitutions that emerge during therapy.[4-8] Variants within the HCV nonstructural protein 5A (NS5A) region substantially increase the risk of virologic failure for many DAA-containing regimens,[9, 10] and there are currently no treatments specifically indicated for either NS5A or NS5B inhibitor–experienced patients.
DAA treatment failure is a growing concern given the long-term persistence of NS5A resistance-associated variants[11, 12] and suboptimal treatment response rates in patients with resistance-associated baseline polymorphisms or treatment-emergent substitutions.[9, 13, 14] Retreatment strategies using 24 weeks of ledipasvir/sofosbuvir have demonstrated a 60% sustained virologic response at posttreatment week 12 (SVR12) rate among patients with baseline NS5A polymorphisms and 33% in patients with NS5A Y93H/N polymorphisms.[14] Similarly, the combination regimen of elbasvir and grazoprevir has reduced efficacy (70%) in GT1a-infected patients with baseline NS5A resistance-associated polymorphisms at elbasvir-specific positions (e.g., M28, Q30, L31, Y93).[9, 15] In addition, the combination of sofosbuvir/velpatasvir has reduced potency to HCV GT1a NS5A variants at position Y93, including the common Y93H variant, which confers a 609-fold increase in half-maximal effective concentration (EC50) to velpatasvir.[16] Therefore, treatments with a high barrier to viral resistance that maintain potency against viral variants, particularly for patients previously treated with HCV DAAs, are needed.
Current recommended retreatment strategies for patients with prior failure of NS3/4A protease inhibitor (PI)–containing DAA regimens include the NS5B nucleotide analogue inhibitor sofosbuvir plus an NS5A inhibitor (ledipasvir, velpatasvir, or daclatasvir) for 12 weeks or the combination of the NS3/4A PI grazoprevir plus the NS5A inhibitor elbasvir with ribavirin (RBV) for 16 weeks.[16] Patients with baseline NS5A variants, either preexisting or the result of treatment emergence from prior exposure to an NS5A inhibitor, have proven more difficult to cure with approved DAA regimens; thus, longer treatment durations, addition of RBV, and the addition of a third or fourth DAA have been examined to maximize SVR12 rates.[14, 17-20]
Glecaprevir (GLE; formerly ABT-493; identified by AbbVie and Enanta) is an HCV NS3/4A PI that has potent pangenotypic antiviral activity. GLE does not inhibit human proteases, exhibits in vitro EC50 values ≤5 nanomolar across all major HCV GTs, and demonstrates <5-fold loss of activity against common GT1 variants at key resistance-associated positions of R155 and D168 to currently available NS3/4A PIs.[6, 21] Pibrentasvir (PIB; formerly ABT-530) is an HCV NS5A inhibitor with EC50 values ≤5 picomolar across all major HCV GTs; it maintains high potency against common NS5A resistance-associated variants, including GT1a Y93H (6.7-fold increase in EC50),[22] which has been associated with reduced susceptibility to other NS5A inhibitors, such as ledipasvir (3,294-fold increase in EC50), daclatasvir (1,600-fold increase in EC50), and velpatasvir (609-fold increase in EC50).[23]
In part 1 of the MAGELLAN-1 study, we evaluated the efficacy and safety of GLE + PIB for 12 weeks, with or without RBV, in patients with prior treatment failure of HCV regimens containing an NS5A inhibitor and/or NS3/4A PI with or without NS5B inhibitors. The impacts of baseline polymorphisms and RBV coadministration on SVR12 rates were also assessed.
Materials and Methods
STUDY OVERVIEW AND REGIMENS
The MAGELLAN-1 (NCT02446717) study was a phase 2, randomized, open-label, multicenter study that assessed the efficacy and safety of GLE + PIB in HCV GT1-infected patients with prior DAA treatment experience. Patients were initially randomized 1:1:1 into three arms (A, B, and C; Fig. 1). Patients enrolled in arm A were treated for 12 weeks with GLE (200 mg once daily) + PIB (80 mg once daily); however, a protocol amendment was implemented to halt enrollment (with 6 patients enrolled) to optimize doses of GLE and PIB for further development.[24] Subsequent to this protocol amendment, the remaining enrolled patients were randomized 1:1 into arm B or C for treatment with GLE (300 mg once daily) + PIB (120 mg once daily) with RBV (arm B; 800 mg once daily) or without RBV (arm C) for 12 weeks. Patients were stratified by HCV subtype (1b or non-1b) and previous DAA class (NS5A inhibitor-experienced, NS3/4A PI-experienced but NS5A inhibitor-naive, or other). For the purpose of analysis, previous HCV treatment experience was deemed cumulative (i.e., a patient exposed to NS3/4A PI and subsequently to NS5A inhibitor was considered NS5A and NS3/4A PI-experienced). All patients signed informed consent, and the study was conducted in accordance with its protocol (designed and sponsored by AbbVie), the Good Clinical Practice Guidelines, and the ethical principles set forth in the Declaration of Helsinki, with independent ethics committee or institutional review board approval for all study sites.
PATIENT POPULATION, CRITERIA, AND STUDY DESIGN
Patients were adults, 18-70 years, without cirrhosis but with chronic HCV GT1 infection and treatment-experienced with a prior DAA-containing regimen. Patients had to have completed past DAA treatment at least 1 month prior to screening visit, with the outcome of prior HCV treatment being either on-treatment virologic failure or posttreatment relapse. Plasma samples for HCV genotyping were collected at screening and assessed with the Versant HCV Genotype Inno LiPA Assay, version 2.0 or higher. The absence of cirrhosis (METAVIR score ≤3, Ishak score ≤4) was determined by one of the following: liver biopsy within 24 months prior to (or during) screening, transient elastography (FibroScan) result of <12.5 kPa within 6 months prior to (or during) screening, or a screening FibroTest score of ≤0.48 with an aspartate aminotransferase to platelet ratio index <1. Patients coinfected with hepatitis B virus, human immunodeficiency virus, or more than one HCV GT at screening were excluded. Key eligibility criteria and definitions of prior treatment responses are provided in the Supporting Information.
EFFICACY, VIROLOGIC, AND SAFETY ASSESSMENTS
Plasma samples for HCV RNA measurements were collected at screening; treatment days 1 and 3 and weeks 1, 2, 4, 6, 8, 10, and 12 (or early discontinuation); and posttreatment weeks 2, 4, 8, 12, and 24 (or early discontinuation) and assessed using the COBAS AmpliPrep/ COBAS TaqMan HCV Quantitative Test, version 2.0. The primary efficacy endpoint was the percentage of patients who achieved SVR12 (HCV RNA <15 IU/mL) in the intent-to-treat (ITT) population, defined as all randomized patients who received at least one dose of study drug. A modified ITT (mITT) analysis was also conducted, excluding all nonvirologic failures (i.e., patients lost to follow-up or early discontinuation). Two-sided confidence intervals (CIs) were determined at a significance level of 0.05 using the Wilson score method for binomial proportions. Statistical summaries were performed using SAS software, version 9.3.
Viral sequences from the baseline plasma sample for each patient were analyzed by next-generation sequencing (Illumina MiSeq) to identify NS3 or NS5A polymorphisms at detection thresholds of 2% and 15%. For patients who had virologic failure within the study, the first samples with HCV RNA ≥1,000 IU/mL collected at or after the time of virologic failure were also analyzed by next-generation sequencing at the same thresholds. Sequences were compared to the corresponding baseline and reference sequences to identify amino acid substitutions that could be associated with resistance to components of the therapy. For HCV resistance analysis, a polymorphism was defined as a baseline amino acid difference relative to the appropriate subtype-specific reference sequence; despite DAA-experienced patients having had prior HCV antiviral therapy, a patient's baseline amino acid variants in this study were considered polymorphisms as the patient's HCV amino acid sequences prior to all previous therapies are unknown. A substitution was defined as a treatment-emergent amino acid sequence different from the patient's baseline viral sequence. An amino acid variant was considered an amino acid change due to a baseline polymorphism or treatment-emergent substitution. Detailed information on the collection of plasma samples, HCV RNA measurement, virologic-failure criteria, and amino acid variants included in resistance analysis are available in the Supporting Information.
Safety assessments were based on the safety population (same as the ITT population). Safety and tolerability assessments were conducted at screening and throughout the study and included monitoring vital signs, physical examinations, adverse events, and clinical chemistry and hematology tests. Adverse events were recorded up to 30 days posttreatment.
Results
A total of 91 patients with HCV GT1 were screened in the United States; 50 were randomized and received at least one dose of study drug. Patients not randomized due to abnormal laboratory values had elevated alanine aminotransferase, aspartate aminotransferase, direct bilirubin, or low platelet count. Among randomized patients, 82% were male, 34% reported black race, and 84% had GT1a infection (Table 1). The majority of patients (66%) had prior treatment failure of regimens containing multiple DAAs; 50% had been previously treated with an NS5A inhibitor, 84% with an NS3/4A PI, and 54% with an NS5B polymerase inhibitor. Of 27 patients with NS5B polymerase inhibitor exposure, 56% had prior treatment with a nucleotide analogue inhibitor, 33% had treatment with non-nucleoside inhibitors, and 11% had exposure to both. The most common prior DAA-containing regimens were boceprevir plus pegylated interferon/RBV (n = 10), telaprevir plus pegylated interferon/RBV (n = 8), ledipasvir/sofosbuvir (n = 8), and simeprevir plus sofosbuvir with or without RBV (n = 8) (Supporting Table S1). Overall, 21/50 patients had prior failure to NS3/4A PI + pegylated interferon/RBV; 5 of those 21 patients were also experienced with NS5A or NS5B inhibitors within the interferon-containing regimen or within other regimens. The type of prior DAA experience (NS3/4A PI only or NS5A inhibitor only or both) was well balanced across treatment groups (Table 1). Next-generation sequencing identified baseline polymorphisms in NS3 and/or NS5A in 86% (43/50) of patients at a detection threshold of 2% and in 80% (40/50) of patients at a threshold of 15%. At a 15% detection threshold, the majority of patients had at least one baseline polymorphism across all treatment groups; although no patients in arm A had polymorphisms in both NS3 and NS5A targets, 27%(6/22) and 41% (9/22) had polymorphisms in both targets in arms B and C, respectively (Table 1). In addition, baseline NS5A polymorphisms were detected in 50% (3/6), 50% (11/22), and 55% (12/22) of patients in arms A, B, and C, respectively, at a detection threshold of 15%. Although a detection threshold of 15% is commonly accepted as clinically relevant, lower thresholds of detection give greater insight into amino acid changes that may emerge in the majority of the population after prior therapy with DAA-containing regimens. At a more sensitive 2% detection threshold, the most common polymorphisms in NS3 were at amino acid positions Q80 (n = 23), R155 (n = 4), D168 (n = 4), and I/V170 (n = 4); and those for NS5A were at positions Q30 (n = 14), Y93 (n = 8), L31 (n = 7), M28 (n = 6), Q54 (n = 6), and H/P58 (n = 5). Baseline amino acid polymorphisms detected at a threshold of 2% are shown in Supporting Table S2, and the prevalence of specific polymorphisms detected at both 2% and 15% is summarized in Supporting Table S3.
Overall, by ITT analysis, SVR12 was achieved in 92% (46/50, 95% CI 81-97) of patients treated with GLE + PIB with or without RBV for 12 weeks. In the halted arm A, in which patients received the lower dose of 200 mg GLE and 80 mg PIB, 100% (6/6, 95% CI 61-100) of patients achieved SVR12. In arm B (300 mg GLE + 120 mg PIB + 800 mg RBV for 12 weeks) SVR12 was achieved in 95% (21/22, 95% CI 78-99) of patients, and among patients in arm C (300 mg GLE + 120 mg PIB for 12 weeks), 86% (19/22, 95% CI 67-95) of patients achieved SVR12 (Fig. 2). The rates of virologic failure were identical (1/22, 5%) with or without administration of RBV (arm B versus arm C). The two patients in arm C who did not achieve SVR12 were lost to follow-up; however, both patients had undetectable HCV RNA at posttreatment week 8. By mITT analysis, excluding patients who failed to achieve SVR12 for nonvirologic reasons, SVR12 rates were 100%, 95%, and 95% for arms A, B, and C, respectively.
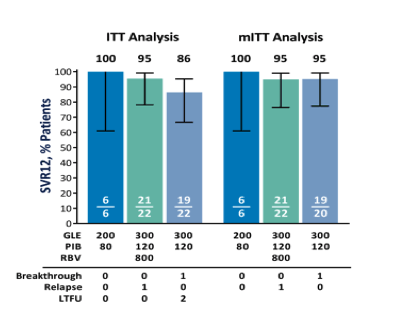
Figure 2.
Sustained virologic response in the ITT and mITT populations. Individual SVR12 rates for arm A (blue), arm B (green), and arm C (gray-blue) are shown for the ITT and mITT populations. The ITT population was all patients that received at least one dose of study drug (n = 50), while the mITT population excluded all patients who did not achieve SVR for reasons other than virologic failure. Whiskers represent the 95% CI using the Wilson score method. Both patients lost to follow-up had nondetectable HCV RNA at posttreatment week 8. Abbreviation: LTFU, lost-to-follow-up.
Two confirmed virologic failures were observed; both patients were compliant and had on-treatment drug levels of GLE and PIB that were consistent with those observed in other patients (Table 2). The patient in arm C (300 mg GLE + 120 mg PIB) had on-treatment HCV breakthrough at week 8; using a 2% detection threshold, this patient had baseline polymorphisms in both NS3 (Y56H and D168A/T) and NS5A (M28V, Q30R, and H58C). At the time of virologic failure, this patient had treatment-emergent substitutions of V36M in NS3 and M28G in NS5A. Of note, although GLE and PIB levels in this patient were within expected therapeutic ranges, the patient had Crohn's disease, was receiving immunosuppressive therapy, and had a prior ileocolectomy. The other patient who had virologic failure was a posttreatment week 4 relapse in arm B (300 mg GLE + 120 mg PIB + 800 mg RBV) and had baseline NS5A polymorphisms L31M and H58D, with no polymorphisms in NS3. At the time of relapse, this patient had treatment-emergent substitutions of A156V in NS3 and Q30R in NS5A. Overall, in patients with baseline polymorphisms in NS3 only, NS5A only, or both NS3 and NS5A at a 15% detection threshold, SVR12 rates were 100% (14/14), 91% (10/11), and 93% (14/15), respectively; SVR12 was achieved by 100% (10/10) of patients with no baseline polymorphisms in NS3 or NS5A.
Adverse events were mostly mild in severity and reported in 84% of patients. Adverse events occurring in ≥10% of patients were headache, fatigue, nausea, and insomnia (Table 3); such events were more common in the RBV-containing arm. Two treatment-emergent serious adverse events were reported, neither of which was deemed related to study drug by the investigator (fractured femur and breast cancer). No patient prematurely discontinued treatment due to adverse events. Clinical chemistry and hematology revealed no significant on-treatment abnormalities in alanine aminotransferase (>3 × upper limit of normal), aspartate aminotransferase (>3 × upper limit of normal), hemoglobin (<10 g/dL), or total bilirubin (>3 × upper limit of normal).
Discussion
The once-daily regimen of GLE and PIB was well tolerated with no serious adverse events related to study drug, no discontinuations due to adverse events, and no relevant laboratory abnormalities. It also resulted in high rates of SVR with or without coadministration of RBV in patients with HCV GT1 infection and prior DAA therapy experience. Overall, the DAA-experienced population in the MAGELLAN-1 study had broad representation of baseline NS3 and NS5A polymorphisms, including polymorphisms at key NS5A positions M28, Q30, L31, H58, and Y93 that confer resistance to earlier-generation NS5A inhibitors. Additionally, all 8 patients previously treated with ledipasvir/sofosbuvir achieved SVR12 despite the presence of the NS5A resistance-associated Y93H/N polymorphism in 5 patients and multiple NS5A polymorphisms in 4 patients. This confirms in vitro data that variants at the Y93 position are susceptible to PIB[22] and suggests GLE plus PIB is an effective treatment for those with a baseline polymorphism or treatment-emergent substitution at this position. Furthermore, 14 of 15 (93%) patients who failed a prior dual (NS3/4A PI plus NS5A inhibitor) or triple (NS3/4A PI plus NS5A inhibitor plus NS5B polymerase inhibitor) DAA regimen achieved SVR12.
The addition of RBV to the GLE + PIB regimen had no apparent impact on response as the rates of virologic failure were identical in arms B and arm C by mITT analysis. All 5 patients who modified RBV dose achieved SVR12, similar to findings with other DAA regimens.[25-27] However, this study did not have a large enough sample size for sufficient statistical power to confirm the impact of RBV on SVR. The patient with virologic relapse in the RBV-containing arm had prior treatment with two different therapeutic regimens: daclatasvir alone and telaprevir plus pegylated interferon with RBV. This patient had L31M and H58D in NS5A at baseline; at the time of virologic failure at posttreatment week 4, a Q30R substitution emerged in this patient in addition to L31M and H58D from baseline. The 1 other patient in this study with a baseline H58D mutation in NS5A also had baseline M28V and Q30R polymorphisms (instead of L31M) in the same target, with an additional Q80K polymorphism in NS3; and this patient achieved SVR12. The patient with virologic breakthrough was on immunosuppressive therapy for Crohn's disease and had prior ileocolectomy; it is unclear whether this contributed to virologic failure. This patient had three NS5A baseline polymorphisms (M28V, Q30R, and H58C) and NS3 baseline polymorphisms at amino acid positions Y56 and D168 that were maintained until virologic failure. While 3 other patients with baseline D168 polymorphisms achieved SVR12, this was the only patient with a baseline Y56 polymorphism. The NS3 V36M and NS5A M28G substitutions emerged in this patient after virologic failure. Both patients who had virologic failure were reported as compliant and had drug exposures similar to other patients.
No clinically significant laboratory abnormalities were reported in hemoglobin levels, including in the RBV-containing arm. Furthermore, no clinically significant chemistry values were observed, including on-treatment elevations in total bilirubin or alanine aminotransferase >3 times the upper limit of normal after alanine aminotransferase normalization or nadir. Although no serious adverse events related to study drug were reported in either arm, the addition of RBV did not reduce the rate of virologic failure but did increase the rate of adverse events. One potential limitation of this study was that RBV coadministration was 800 mg daily, regardless of patient weight, i.e., below the conventional 1,000 or 1,200 mg weight-based RBV dosing. Because of this, it is unclear whether weight-based RBV coadministration could have increased the efficacy in arm B. By corollary, it is also likely that a conventional weight-based RBV dose would have resulted in a further increased rate of adverse events for these patients when compared to that seen with the lower dose of RBV in arm B as increasing side effects with higher RBV dose is well documented.[28] However, regardless of comparison between treatment arms, the low rate of virologic failure in the RBV-sparing arm suggests that the response rate is already near maximal, and the addition of RBV may not impact this. The patient population enrolled here is considered inherently difficult to cure, owing to increased prevalence of baseline resistance-associated polymorphisms likely stemming from prior DAA failure.[15, 29-32] As such, additional study is required in this diverse patient population to further confirm efficacy of the regimen in GT1 patients with prior failure of DAA therapy, including patients with prior exposure to DAA-containing therapies and concomitant cirrhosis, who were excluded here.
In summary, the combination of GLE and PIB showed potent antiviral activity, regardless of the presence of one or more baseline resistance-associated polymorphisms and irrespective of previous DAA-containing treatment regimens, resulting in high SVR12 rates in patients without cirrhosis but with HCV GT1 infection. This suggests that the combination of GLE and PIB is highly effective in this population, which currently has limited treatment options. Based on these findings, larger and more diverse patient groups are being evaluated in phase 3 studies to confirm the safety and efficacy of the RBV-free coformulation of GLE/PIB (300 mg/120 mg) in all six major HCV GTs, including patients with prior DAA experience and compensated cirrhosis.
|
|
| |
| |
|
|
|