 |
|
|
| |
New 2006 Recommendations of the International AIDS Society-USA Panel
|
| |
| |
JAMA. Aug 16 2006;296:827-843.
In Toronto at IAS, Scott Hammer and colleagues present updated guidelines from the International AIDS Society - USA panel for starting antiretroviral therapy, the regimen to use, monitoring of patient response, and circumstances that should prompt changes in therapy.
Scott M. Hammer, MD; Michael S. Saag, MD; Mauro Schechter, MD, PhD; Julio S. G. Montaner, MD; Robert T. Schooley, MD; Donna M. Jacobsen, BS; Melanie A. Thompson, MD; Charles C. J. Carpenter, MD; Margaret A. Fischl, MD; Brian G. Gazzard, MA, MD; Jose M. Gatell, MD, PhD; Martin S. Hirsch, MD; David A. Katzenstein, MD; Douglas D. Richman, MD; Stefano Vella, MD; Patrick G. Yeni, MD; Paul A. Volberding, MD
The International AIDS Society-USA panel has published its antiretroviral therapy guidelines 7 times since 1996, a period that coincides with the rapid evolution in practice brought on by the HAART era. The rationale for issuing revised guidelines in 2006 is based on several developments: (1) continued refinement of the recommended initial treatment regimen with a focus on the nucleoside (or nucleotide) reverse transcriptase inhibitor (nRTI) components of nonnucleoside reverse transcriptase inhibitor (NNRTI)- and protease inhibitor (PI)-based regimens; (2) the US Food and Drug Administration (FDA) approvals of tipranavir and darunavir, which provide new options for management of treatment-experienced patients; (3) the redefinition of the goal of regimens for highly treatment-experienced patients to achieve plasma human immunodeficiency virus 1 (HIV-1) RNA levels below assay detection limits; (4) the availability of a triple-drug combination formulated in 1 pill to be given once daily; and (5) new information on drug-sparing therapeutic strategies, such as supervised treatment interruptions and ritonavir-boosted PI monotherapy.
"...treatment interruption for successfully treated patients is not recommended outside of clinical trials- In the setting of treatment experience, resistance testing should be performed while the patient is taking the failing regimen. -- Trials with newer antiretroviral agents have shown that it is possible to achieve plasma HIV-1 RNA levels below 50 copies/mL even in highly treatment-experienced patients- If at least 2 drugs cannot be identified, strong consideration should be given to maintaining the current regimen until new drugs become available, assuming immunologic and clinical stability -... If several potent drugs other than enfuvirtide are available, it may be best to defer enfuvirtide use until it becomes 1 of 2 available and fully active drugs (AII). However, since the goal of therapy is to achieve plasma HIV-1 RNA levels of less than 50 copies/mL, enfuvirtide often is required to achieve this degree of success among heavily antiretroviral-experienced patients-... Resistance assays may also be of value in selecting the initial treatment regimen, because transmission of drug-resistant HIV strains leading to suboptimal virologic responses... For patients without symptoms, therapy should be initiated at some point after the CD4 cell count declines below 350/μL but before it reaches 200/μL...:
On the horizon are investigational antiretroviral drugs in existing classes such as the NNRTIs etravirine and TMC-278, as well as drugs in novel classes. The development of CCR5 inhibitors illustrates the complexity and unpredictability of antiretroviral agent development. For example, CCR5 coreceptor antagonists have encountered challenges. Aplaviroc's development was stopped because of hepatotoxicity. Vicriviroc's development in treatment-naive patients was discontinued because of unexpected virologic failures and the drug is being carefully scrutinized to determine if lymphoma development is potentiated by the drug in treatment-experienced persons. Maraviroc's development in both treatment-naive and treatment-experienced persons is ongoing. Encouragingly, integrase inhibitors (MK-0518 and GS-9137) are showing promise,132-133 and proof-of-principle for the maturation inhibitor, PA-457, has been demonstrated in humans.134 It is quite possible that paradigms of treatment will be altered by 1 or more of these agents-that is, when to start therapy and with what, may well change in the years ahead.
Either of 2 basic 3-drug regimens continues to be recommended for initial therapy: NNRTI-based or PI ritonavir-boosted-based combinations. Of the NNRTIs, efavirenz is recommended due to its consistent efficacy demonstrated in numerous randomized trials and its toxicity profile
Of the ritonavir-boosted PIs, recommended components are lopinavir (AIa), atazanavir (BIII), fosamprenavir (BIII), or saquinavir (BIII)... Either of 2 basic 3-drug regimens continues to be recommended for initial therapy: NNRTI-based or PI ritonavir-boosted-based combinations. Of the NNRTIs, efavirenz is recommended due to its consistent efficacy demonstrated in numerous randomized trials and its toxicity profile ...... Recommended nRTI components in the initial regimen are tenofovir and emtricitabine (AIa), zidovudine and lamivudine (AIa), or abacavir and lamivudine (AIa). Tenofovir is well tolerated but should be used with caution, or avoided, in patients with preexisting renal insufficiency (AIa).
When to Change and What to Change
Recent Data
Despite availability of regimens that are potent, well tolerated, convenient, and relatively easy to take, many patients still require a change in regimen, often related to treatment-related toxic effects, intolerance, inconvenience, or failure. Trials with newer antiretroviral agents have shown that it is possible to achieve plasma HIV-1 RNA levels below 50 copies/mL even in highly treatment-experienced patients.66-67 Similarly, several studies have demonstrated that many treatment-related toxic effects can be avoided, reversed, or at least partially controlled with judicious modifications of antiretroviral regimens.68-69
Changing for Treatment Failure
The benefits of plasma HIV-1 RNA suppression to less than 50 copies/mL on durability of response and prevention of emergence of resistance support using persistent elevations above this cutoff as a definition of virologic failure. Previous guidelines recommended establishing a plasma HIV-1 RNA target of at least 0.5 to 1 log10 HIV-1 RNA copies/mL below baseline for patients with more advanced treatment failure and a high level of multidrug resistance. However, several recent studies evaluating newer antiretroviral agents designed to have activity against multidrug-resistant virus have demonstrated that a high proportion of heavily treatment-experienced patients can achieve HIV-1 RNA levels of less than 50 copies/mL.66, 72-73 When this is not achievable, stability of CD4 cell count and clinical status usually can be maintained for relatively long periods with reductions of HIV-1 RNA to levels at least 0.5 to 1.0 log10 copies/mL below baseline, although cumulative acquisition of new resistance mutations is a consequence of this approach. Isolated episodes of intermittent viremia or transient episodes of plasma HIV-1 RNA levels higher than 50 copies/mL but lower than 500 to 1000 copies/mL do not necessarily predict subsequent virologic failure and should not prompt an immediate change in therapy.74
For selecting subsequent therapy, data from recent trials showed no benefit of double-boosted PIs (2 active PIs and low-dose ritonavir) over single-boosted PIs.66, 72 Moreover, pharmacokinetic interactions, tolerance, and long-term adverse effects complicate double-boosted PI therapy.
Drug-Sparing Strategies
Ritonavir-Boosted PI Monotherapy. The potency and high genetic barrier to resistance of ritonavir-boosted PIs might make them potentially useful as initial therapy or as part of a simplification strategy. In the OK study, 42 participants were randomly assigned to continue lopinavir and ritonavir plus 2 nRTIs or to begin lopinavir and ritonavir monotherapy following suppression to less than 50 copies/mL on lopinavir and ritonavir plus 2 nRTIs. At 48 weeks, 81% and 95% of the participants in the 2 groups, respectively, maintained HIV-1 RNA levels lower than 50 copies/mL. This was not statistically different because the numbers in the trial were small.75 In the single-group, pilot ACTG A5201 study76 of 36 participants, simplification of therapy to atazanavir and ritonavir alone following 6 weeks of induction with atazanavir and ritonavir plus 2 nRTIs resulted in a 91% rate of suppression of plasma HIV-1 RNA to less than 50 copies/mL at 24 weeks. In both of these studies, PI mutations were not detected in the patients in whom monotherapy failed virologically. These data are still preliminary but add to the growing experience with nRTI-sparing strategies and suggest that further studies are warranted.
Treatment Interruptions and Intermittent Therapy.
Recent studies have demonstrated no beneficial effect, and sometimes deteriorating clinical outcomes, by using structured (or supervised) treatment interruptions (STIs) as a treatment strategy. Two general approaches have been evaluated: therapy interruption done at predefined intervals of time or interruption based on targeted CD4 cell responses.
The Staccato,77 Window,78 Trivacan,79 and Istituto Superiore di Sanita Pulsed Anti-Retroviral Therapy (PART)80 studies each looked at variable or fixed intervals of treatment interruptions that lasted from weeks to months. A common theme that emerged is that short intervals of stopping and starting therapy can be associated with relatively high rates of emergence of drug resistance and are generally not advisable. Longer intervals of starting and stopping therapy may not result in significantly more failure, but there is no clear consensus on the safety and value of this approach.
Other studies used CD4 cell count triggers of treatment interruption. The Strategies for Management of Anti-Retroviral Therapy study81 evaluated routine interruption of therapy when CD4 cell counts reached a threshold of more than 350/μL and reintroduction of therapy when the CD4 cell count decreased to less than 250/μL vs continuous therapy. There was increased progression to a new AIDS-defining event or death, as well as more non-HIV-related serious adverse events among those in the STI group than those in the control group. Similarly, the group whose treatment was interrupted by CD4 cell count in the Trivacan study was discontinued early as a result of an increased incidence of severe morbidity.78
Available data from ongoing trials evaluating treatment interruptions at higher CD4 cell counts are difficult to interpret, given their lack of statistical power to compare clinical end points.77, 80, 82
Some studies that used STI as a means to improve immunologic host responses to HIV through autoimmunization have shown some improvement in the level of HIV-1 viremia in the STI group compared with the control group. In one such study, the effect of the STI was small and several participants developed mutations associated with resistance to the drugs in their discontinued regimens and had difficulty reestablishing control of replication when therapy was reintroduced.83
New Drugs
Since the previous guidelines, 2 new drugs have become available for use in treatment-experienced patients.
Tipranavir. A PI designed to have activity against multi-PI-resistant virus is the combination of 500 mg of tipranavir with 200 mg of ritonavir twice a day. The Randomized Evaluation of Strategic Intervention in Multi-drug Resistant Patients with Tipranavir (RESIST) trials I84 and II75, 85 evaluated tipranavir in patients in whom PI-, NNRTI-, and nRTI-containing regimens had failed. Patients who had 2 or more mutations associated with high-level tipranavir resistance were not eligible for enrollment. The 2 studies had similar inclusion criteria and were conducted in North America, Europe, Australia, and Latin America. The tipranavir-plus-ritonavir group demonstrated greater reductions in plasma HIV-1 RNA levels and increases in CD4 cell counts than did the comparator PI group when each was combined with optimized background regimens. Of note, in this heavily treatment-experienced population, 33% of the enfuvirtide-naive participants achieved levels of less than 50 copies/mL if enfuvirtide was part of the optimized background regimens.66 The likelihood of reaching an HIV-1 RNA level lower than 50 copies/mL was highest if more than 2 active drugs were in the regimen, especially if 1 of the drugs was enfuvirtide.
The principal toxic effects of tipranavir are gastrointestinal, with approximately 20% of participants experiencing nausea and 30% diarrhea. Fatal and nonfatal intracranial hemorrhage has recently been reported among patients taking tipranavir. Liver enzyme elevations are not uncommon, particularly in patients with chronic HBV or HCV, or with elevated liver enzymes at initiation of tipranavir-ritonavir therapy. As with other PIs, the presence of a greater number of key mutations leads to stepwise reduction in activity.88 A 1.8-fold phenotypic change in tipranavir susceptibility is associated with reduced activity of tipranavir-ritonavir-containing regimens in the clinical studies conducted thus far. Pharmacologic interactions between tipranavir and other drugs metabolized by the liver may limit its use for patients taking numerous drugs.
Darunavir. Formerly known as TMC-114, darunavir, is a potent new PI also designed to have activity against multi-PI-resistant virus; it is given at a dose of 600 mg twice a day combined with low-dose ritonavir (100 mg twice a day). Darunavir was evaluated in 2 studies (Performance of TMC-114/r When Evaluated in Treatment-Experienced Patients With PI Resistance [POWER] I and II)86-87 conducted in the United States, Europe, and South America. Entry criteria were similar to that in the RESIST trials84-85 except that the existence of baseline genotypic mutations associated with PI resistance was not an exclusion criterion. Heavily treatment-experienced participants (eg, there was a 70- to 80-fold reduction in lopinavir susceptibility) were assigned to receive optimized background regimens combined with either darunavir or a comparator PI. The comparator PI subset performed similarly as it had in the RESIST studies. Plasma HIV-1 RNA level reductions and CD4 cell count increases were significantly greater in the darunavir group. Of note, at 24 weeks more than 60% of enfuvirtide-naive participants who were treated with enfuvirtide achieved undetectable levels.74 POWER III,67 an open-label study, provided safety and relative efficacy data for darunavir and ritonavir in heavily treatment-experienced patients.
The principal toxic effects of darunavir are gastrointestinal, 18% of participants experienced nausea and 17% diarrhea. Mutations at positions 32, 33, 47, and 54 on the protease gene are associated with reduced susceptibility to darunavir and a 4- to 10-fold change in phenotypic susceptibility is associated with reduced activity of drug.89
Recommendations
The recommendations for when to change and what to change to depend on the reasons for changing and on the availability of active drugs for constructing a potent regimen.
Changing Therapy Because of Toxicity, Intolerance, or Inconvenience. Low-grade and often transient symptoms that typically occur early after initiation of therapy (eg, zidovudine-related headache and nausea; efavirenz-related central nervous system adverse effects) can often be addressed with appropriate patient education and symptomatic medications without stopping the offending drug.
In successfully treated patients who need to modify their regimens because of toxic effects or intolerance and in whom the offending agent can be identified with reasonable certainty, single-drug substitution is generally safe, particularly in previously treatment-naive patients not expected to be harboring archived drug-resistance mutations (AII).
When a toxic effect cannot be confidently attributed to a single drug and is severe enough to require temporary discontinuation of therapy, all drugs in the regimen should be stopped (AII).
For patients taking drugs with substantially different half-lives (eg, NNRTI and nRTIs) and whose reason for changing therapy is inconvenience or adverse effects that do not require immediate action, staggered discontinuation of the drugs should be considered (eg, stopping the NNRTI 5 to 7 days before the nRTIs), in an attempt to avoid the emergence of drug resistance (BIII). Nevertheless, given wide interpatient variability, it is not possible to determine with certainty what is a safe time interval for differential stoppage of antiretrovirals.90 Once the toxicity resolves, a new regimen can often be introduced.
Symptomatic lactic acidosis is a life-threatening condition that is most often associated with the use of nRTIs, particularly with stavudine.91 Immediate discontinuation of the antiretroviral regimen is indicated. Following complete recovery, the safest course is to introduce an nRTI-sparing regimen, such as a ritonavir-boosted PI with an NNRTI (BII). However, nRTIs less commonly associated with mitochondrial toxicity, such as lamivudine, emtricitabine, tenofovir, and abacavir, may be safely reintroduced following full recovery from this syndrome if the benefit is thought to outweigh the risk. Close monitoring of symptoms and lactate levels is required if this is attempted (BIII). Of note: routine lactate monitoring in an asymptomatic individual who has not experienced an episode of lactic acidosis is not recommended.
In the case of hyperlipidemia, adjustments in the antiretroviral regimen are recommended as a primary approach if diet and exercise fail to control lipid levels (AI).68-70 For patients who do not want to change their regimen, the addition of a lipid-lowering agent is an acceptable strategy (AII). If other viable agents with presumptive antiretroviral activity are not available, then it is more appropriate to add specific antilipid therapy (AI). When there are changes in body fat distribution, particularly lipoatrophy, switching the putatively offending antiretroviral agent(s) may halt further progression of the body-shape changes and, in some cases, can lead to some degree of reversal of the abnormality over an extended period (AI).69 However, selection of the next regimen often poses management challenges because a number of drugs from different classes are associated with lipodystrophy. Given that replacing the drug(s) responsible usually does not completely reverse the abnormality, close monitoring for the first signs of body fat changes and early switching, if options exist, are recommended (AII).92
Changing Therapy Because of Treatment Failure. Treatment failure may be defined virologically, immunologically (declining CD4 cell count), or clinically (HIV-related disease progression). Viral rebound should be confirmed to ensure that it is not transient (ie, a blip).
FIRST REGIMEN FAILURE. The fundamental principle for managing any regimen failure, regardless of how many prior regimens the patient has experienced, is to ensure that at least 2, and preferably 3, drugs used in the new regimen are likely to have activity based on integration of resistance test results and history of antiretroviral regimen use. In individuals in whom the first regimen fails and who were likely infected with a drug-susceptible virus, a full assessment of adherence is the first step. Particular attention should be paid to subtle toxic effects, such as low-grade nausea or headache, which may interfere with optimal adherence. If attempts at improving adherence fail and plasma HIV-1 RNA levels are confirmed to be higher than 500 to 1000 copies/mL, resistance testing should be obtained. Full susceptibility to all drugs in the regimen suggests that the patient is not taking the drugs. If drug resistance is detected, the regimen should be altered so that there are at least 2 fully active drugs in the regimen.
MULTIPLE REGIMEN FAILURE. In the setting of 3 or more regimen failures, management challenges increase substantially. Subsequent regimen failures cause further drug resistance that limits the remaining antiretroviral options. A crucial concept when initiating a new regimen after treatment failure is the requirement of preferably 3, but at least 2, fully active agents as determined by resistance test results and prior treatment history (AI). If at least 2 drugs cannot be identified, strong consideration should be given to maintaining the current regimen until new drugs become available, assuming immunologic and clinical stability (AI). Investigational drugs often become available through clinical trials and physicians should be vigilant for drugs in development that may become available. The use of a single-active drug, so-called sequential monotherapy, should be avoided since it usually leads to rapid development of resistance to that drug, further limiting future treatment options (AI). When 2 or more potent drugs are identified, the goal of therapy should be the achievement of HIV-RNA levels below 50 copies/mL, even for highly treatment-experienced patients (AI). If durable undetectable levels of HIV-1 RNA are deemed unachievable, the goal of therapy shifts to maintenance of immunologic integrity and prevention of clinical disease progression with acceptance of incomplete viral suppression (AII).
When choosing the next regimen, maintenance of nRTI agents in the regimen still contributes some antiviral activity, even when formal resistance is detected.93 In particular, lamivudine or emtricitabine often continues to have significant activity (0.5 to 0.8 log10-copies/mL declines) even when resistance-conferring mutations to these drugs are present (eg, the M184V or L44I substitutions).94 In contrast, currently available NNRTIs typically have no virologic activity when high-level resistance is demonstrated and should not be continued in the next treatment regimen (AI).
There are no convincing data to support the use of a double-boosted PI and these combinations should be avoided (AI).
The RESIST66 and POWER67 studies have helped define the optimal time in which to use enfuvirtide. If several potent drugs other than enfuvirtide are available, it may be best to defer enfuvirtide use until it becomes 1 of 2 available and fully active drugs (AII). However, since the goal of therapy is to achieve plasma HIV-1 RNA levels of less than 50 copies/mL, enfuvirtide often is required to achieve this degree of success among heavily antiretroviral-experienced patients (AI). There are limited and somewhat conflicting data on the potential benefit of maintaining enfuvirtide in the regimen during virologic failure. In these patients, enfuvirtide-resistant virus is frequently present. Although resistance to enfuvirtide may be associated with decreased viral replicative capacity, its removal from the regimen may not lead to significant drops in CD4 cell count.95 Thus, given its cost and inconvenience, consideration should be given to stopping enfuvirtide in case of virologic failure (BIII); additional data are needed to guide clinicians with this decision.
Discordant Responses. Some patients experience a discordant response, whereby the HIV-1 RNA level is below the limit of detection but the CD4 cell count response is blunted. In such settings, it is prudent to continue the current regimen (AII). Changing or intensifying the regimen has not been shown to have an effect on the CD4 cell count response, except in the case of patients using drugs that are associated with lymphopenia (zidovudine or didanosine; AII). The use of interleukin-2 may cause significant toxic effects and no clinical benefit has yet been documented; thus, it should not be used except in clinical trials (AIII). Other patients may exhibit a different pattern of discordant response, characterized by a sustained CD4 cell count response, despite persistent viremia. Both types of discordant responses, particularly the former, have been associated with rates of progression to AIDS or death that are intermediate between those observed in complete responders and in nonresponders.96-98
Treatment Interruptions and Intermittent Therapy. At present, a treatment interruption for successfully treated patients is not recommended outside of clinical trials (AI). Cycles of STI in patients with controlled viremia simply to reduce long-term exposure to the drugs are also not recommended (AII). Similarly, STI to allow reversion to wild-type virus before instituting a new regimen is not recommended because this approach has not been demonstrated to be beneficial and may be detrimental (AI).
The remaining situations in which STI can still be considered are in cases of significant antiretroviral toxic effects (AIII) and for the treatment of intercurrent infections in situations in which significant drug interactions might jeopardize the efficacy of either treatment (AII). Treatment fatigue, when a patient strongly requests that treatment be stopped temporarily (AII) is a common reason to consider an STI.
Antiretrovirals should be reinstituted once the toxicities resolve, the infection has been treated, or the patient is ready to restart treatment (BII). In the case of STI for treatment fatigue, the patient should be counseled about the risk of possible disease progression and the risk of drug resistance once therapy is stopped. If the STI is instituted, close monitoring is advised.
Resistance Testing
Genotypic and phenotypic assays are widely used to evaluate HIV resistance to antiretroviral drugs.60 In the setting of treatment experience, resistance testing should be performed while the patient is taking the failing regimen. Resistance assays may also be of value in selecting the initial treatment regimen, because transmission of drug-resistant HIV strains leading to suboptimal virologic responses has been documented and there is evidence of increasing rates of drug resistance among newly diagnosed patients both in Europe and the United States.61 Early virologic failure in patients receiving combination antiretroviral therapy has been shown to be often associated with resistance to a single component of a multidrug regimen.62-63
Results for genotypic assays can be available in 1 to 2 weeks, whereas results for phenotypic assays can take 3 to 4 weeks; however, interpretation may be complex, requiring precise knowledge of the mutations associated with decreased susceptibility to each antiretroviral drug, the interactions among these mutations, and their potential to confer cross-resistance.64 Expert advice should be sought whenever possible to interpret genotypic results.
Phenotypic assays quantify the ability of the virus to grow in varying concentrations of specific antiretroviral drugs. Automated recombinant virus phenotypic assays are commercially available; however, the test usually requires weeks for turnaround and is more expensive than genotypic testing.
Cost, differing interpretations of resistance testing, and insensitivity for detection of minor viral species are limitations of genotypic and phenotypic tests. Both tests identify only the predominant circulating virus in plasma (dependent on the selective pressure exerted by the patient's current regimen).
Recommendations
Once antiretroviral therapy is initiated, plasma HIV-1 RNA level should be checked relatively frequently (eg, every 4-8 weeks; AIIa) until it reaches levels below the limits of detection of the assay and regularly thereafter (eg, 3 times per year to 4 times per year [BIII]).65 CD4 cell count generally should be monitored in tandem with HIV-1 RNA level.
Genotypic testing for HIV resistance is preferred over phenotypic testing in most settings because it is faster, readily available, and less expensive; phenotypic testing may be more useful for patients with virologic failure following 2 or more regimens. Baseline resistance testing is recommended if the prevalence of transmitted HIV drug resistance is greater than 5% (BIII) and should be considered if the prevalence is unknown, but antiretroviral penetration in the population is thought to be high enough that transmission of drug resistance is likely (BIII). Thus, resistance testing has become a routine part of the baseline evaluation of patients with established infection in many settings.7 Resistance testing is recommended in the setting of virologic failure (AIa) and ideally should be performed when the patient is taking the failing regimen, which maximizes selective pressure on the virus thus increasing the likelihood that resistance testing will detect the mutations that the patient harbors. Resistance testing should also be considered after a new regimen is introduced if the HIV-1 RNA trajectory is not optimal (AII). Drug-resistance testing should not be performed if the plasma HIV-1 RNA level is below 500 to 1000 copies/mL because the assay does not perform reliably at that level.
Therapeutic drug monitoring for NNRTIs and PIs has entered clinical practice in a number of countries (eg, Western Europe and Canada) but in others remains a research tool and is not recommended as a part of routine care (CIII). Monitoring of serum nRTI concentrations is not recommended in clinical practice (CIII). Replication-capacity assays are also not recommended as part of routine care, although, as noted, this in vitro viral characteristic is already being reported by at least 1 commercial drug-resistance testing company (CIII).
Antiretroviral therapy in adults continues to evolve rapidly, making delivery of state-of-the-art care challenging. Initiation of therapy continues to be recommended in all symptomatic persons and in asymptomatic persons after the CD4 cell count falls below 350/μL and before it declines to 200/μL. A nonnucleoside reverse transcriptase inhibitor or a protease inhibitor boosted with low-dose ritonavir each combined with 2 nucleoside (or nucleotide) reverse transcriptase inhibitors is recommended with choice being based on the individual patient profile. Therapy should be changed when toxicity or intolerance mandate it or when treatment failure is documented. The virologic target for patients with treatment failure is now a plasma HIV-1 RNA level below 50 copies/mL. Adherence to antiretroviral therapy in the short-term and the long-term is crucial for treatment success and must be continually reinforced.
The guidelines are internationally based and designed for caregivers who practice in relatively resource-unconstrained environments with regard to the availability of drugs and monitoring tools. However, the principles of therapy outlined in these guidelines are pertinent to antiretroviral rollout initiatives in resource-limited settings in that key to the success of such programs are use of potent combinations of drugs designed to fully suppress virus replication, excellent adherence, and avoidance of toxicity.12 As drugs and diagnostic tools become more affordable and widely available, the principles outlined herein can help guide national programs in the developing world.
Strength of Recommendation and Quality of Evidence Rating Scale
Strength of Recommendation
A: Strong evidence to support the recommendation
B: Moderate evidence to support the recommendation
C: Insufficient evidence to support the recommendation
Quality of Evidence*
Ia, Ib: Evidence from 1 or more published randomized, controlled clinical trials
IIa, IIb: Evidence from nonrandomized clinical trials; cohort or case-control studies
III: Recommendation based on the panel's analysis of the accumulated available evidence
*"a" Indicates published in the peer-reviewed literature and "b" indicates presented in abstract form at peer-reviewed scientific meetings.
When to Start Antiretroviral Therapy
Antiretroviral therapy is recommended for all patients with symptomatic HIV disease (AIa; Table 1).13 For patients without symptoms, therapy should be initiated at some point after the CD4 cell count declines below 350/μL but before it reaches 200/μL (AIIa). No new evidence has emerged to define the optimal CD4 cell count that provides a treatment-related survival advantage, and based on the inherent difficulty with designing and executing such studies, it is unlikely that a randomized, controlled trial will be conducted to answer this question. Rather, recommendations rely on well-conducted cohort studies.18 Data from one observational study showed a benefit to starting therapy when CD4 cell counts were higher than 350 cells/μL compared with starting at an unspecified later time, but these data do not resolve the questions of the precise CD4 cell count at which to start.19 Even when CD4s are 350-500 & you see in table just below 'treatment generally not recommended' it says in legend 'consider treatment if viral load is >100,000 copies/ml or with rapid decline in CD4'.
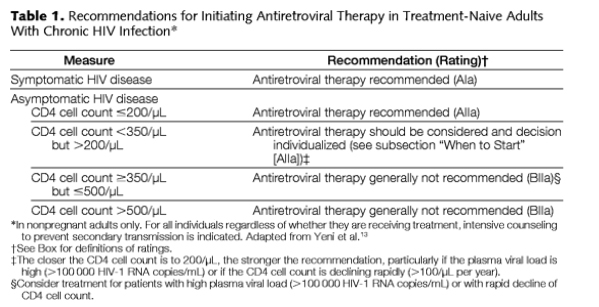
Individualization continues to guide the timing of treatment initiation, with consideration of patient readiness, rate of CD4 cell count decline, and plasma HIV-1 RNA level.13 Newer formulations of antiretroviral drugs and combinations with improved tolerance and convenience may mitigate previous reluctance to begin therapy early.
The debate about aggressive antiretroviral treatment of primary (acute) HIV infection continues. Recent reports of substantial depletion of CC chemokine receptor 5 (CCR5)-expressing CD4 cells in gut-associated lymphoid tissue in the setting of primary infection, which may be slow or refractory to reconstitution with antiretroviral therapy, represent advances in the understanding of HIV pathogenesis that confirm earlier studies in the simian immunodeficiency virus-rhesus macaque system.20-23 It remains to be determined what the implications are for the timing of therapy in established HIV infection.
Choice of Initial Regimen
Recent Data
Since the last edition of these guidelines, clinical trial and cohort studies have led to refinements in the choice of initial regimen. The recommended initial regimen remains a combination of 2 nRTIs with either an NNRTI or a PI boosted with low-dose ritonavir. Given the high degree of comparability of the recommended components of these regimens in treatment-naive persons with drug-susceptible virus, the choice of drug centers on acceptability; predicted tolerance; pill burden; comorbid conditions; short-term, mid-term, and long-term adverse event profiles; and successful alternatives should the initial regimen fail and drug resistance emerge. The successful outcomes of several "switch studies" suggest that the initial choice of regimen does not preclude safely changing drugs once viral suppression is achieved.
There are no data to establish that either an NNRTI- or a PI/ritonavir-based regimen is superior in efficacy at any stage of disease. Although some providers prefer a ritonavir-boosted PI over an NNRTI in very advanced disease with high viral loads because of the higher genetic barrier to resistance and slower rate of mutation selection seen with PIs, data to support PI- over NNRTI-based regimens are not yet reported. They perform equally well in settings with low CD4 cell counts and high plasma HIV RNA levels.
Specific considerations in selecting an initial regimen include results of baseline genotypic drug resistance testing; viral hepatitis coinfection status; presence of lipid abnormalities, diabetes mellitus, cardiac, hepatic, or renal dysfunction; reproductive status and use of contraception; and concomitant medications (see the "Special Populations" subsection).
NNRTI-Based Regimens. Data continue to accrue confirming the efficacy and ease of NNRTI-based regimens. Efavirenz plus 2 nRTIs has become a standard-of-care comparator in clinical trials. Efavirenz use requires adequate contraception in women of child-bearing potential given its teratogenic risk in the first trimester. Efavirenz is available in a fixed-dose formulation with tenofovir and emtricitabine, which allows patients to take only 1 pill a day.
Nevirapine has virologic activity similar to efavirenz and is safe for the fetus in all stages of pregnancy if appropriate for the mother. There is a risk of potentially fatal hepatotoxicity in women with CD4 cell counts higher than 250/μL and in men with counts higher than 400/μL. Data from Thailand,24 where the drug was well tolerated in the patients studied, have raised the question about the true risk to women and whether it applies to all populations. It is not known whether switching from efavirenz to nevirapine when the CD4 cell count has risen above these thresholds while receiving treatment is associated with the same risk as starting nevirapine de novo in a treatment-naive woman or man with CD4 cell counts higher than 250 or 400 cells/μL, respectively.
Since the last Guidelines publication, data confirm that 4 drugs are generally no better than 3 drugs when considering treatment with currently available nRTIs and PIs in treatment-naive patients not infected with drug-resistant virus.25 In the final analysis of AIDS Clinical Trials Group (ACTG) A5095, which had previously shown that the zidovudine, lamivudine, and abacavir regimen was inferior to efavirenz plus 2 nRTIs,26 efavirenz plus zidovudine and lamivudine performed comparably to efavirenz plus zidovudine, lamivudine, and abacavir with 80% and 86% rates of virologic suppression to less than 50 HIV-1 RNA copies/mL of plasma at 48 weeks, respectively.27
PI-Based Regimens. Ritonavir-boosted PIs remain a standard-of-care option for initial treatment.13
The largest cumulative data set exists for ritonavir-boosted lopinavir, which now is available in a formulation that does not require refrigeration. This is especially important for the developing world. To the developed world, it offers convenience and lower pill burden. Since our last report, ritonavir-boosted lopinavir has been approved for once daily dosing in treatment-naive patients in the United States. Approvals in other countries are pending. The incidence of diarrhea is greater with 800 mg of ritonavir with 200 mg of lopinavir given once a day than with 400 mg of ritonavir with 100 mg of lopinavir twice a day. Lipid elevations appear comparable. The single dosage of ritonavir-boosted lopinavir is not approved for treatment-experienced persons or for children.
Direct comparative trials of ritonavir-boosted fosamprenavir or atazanavir vs lopinavir in treatment-naive patients have not been formally reported; however, results of a study of the latter comparison (the KLEAN trial) are expected to be released in August 2006. Ritonavir-boosted atazanavir has the potential advantage of causing less hyperlipidemia than other ritonavir-boosted PIs.28 Its major drug-specific adverse effect is hyperbilirubinemia, which is more frequent in persons with the uridine 5'diphospho-glucuronosyl transferase (UGT) UGT1A1-28 genotype or the CC genotype of the 3435CT polymorphism in the multidrug resistance (MDR1) gene.29-31 The latter is associated with higher atazanavir levels and thus a greater risk of hyperbilirubinemia. These genotypic associations, along with the HLA-B-5701 genotype associated with abacavir hypersensitivity, may ultimately lead to a greater degree of individualization of therapy with routine genotypic profiling of patients in the future. However, it is premature to recommend this form of patient screening. With atazanavir in particular, the indirect hyperbilirubinemia is generally asymptomatic and unassociated with liver enzyme elevations. Knowledge of an increased risk of hyperbilirubinemia would not necessarily preclude its use.
Resistance patterns at the time of virologic failure among participants in the Bristol-Myers Squibb AI424-089 trial32 support the use of ritonavir-boosted atazanavir over atazanavir alone when choosing this agent as part of an initial regimen. In this study, 200 treatment-naive participants were randomized to receive atazanavir or atazanavir plus ritonavir each in combination with extended-release stavudine and lamivudine. In the intent-to-treat analysis, at 48 weeks, HIV-1 RNA suppression to levels less than 50 copies/mL was comparable: 70% and 75%, respectively. Of the 10 virologic failures in the atazanavir-alone group, PI mutations were seen in 3 and nRTI mutations in 10 participants. Of the 3 virologic failures in the group receiving atazanavir plus ritonavir, no PI mutations and 1 nRTI mutation were seen. Hyperbilirubinemia and lipid elevations were greater in the group receiving atazanavir plus ritonavir.
In the MaxCmin2 trial,33 400 mg of lopinavir and 100 mg of ritonavir twice daily plus 2 nRTIs was compared with the soft-gel formulation of 1000 mg of saquinavir plus 100 mg of ritonavir twice daily plus 2 nRTIs in treatment-naive participants. At 48 weeks, the virologic failure rate was higher in the saquinavir-plus-ritonavir group. This difference may have occurred because patients had less tolerance for the saquinavir-based combination than those taking the lopinavir-based combination. A trial with the saquinavir hard-gel formulation showed good virologic efficacy.34
Dual nRTI Components. Nonnucleoside reverse transcriptase inhibitors or PIs typically are given with 2 nRTIs in combinations that have advantages with respect to side effect profiles and availability in fixed-dose combinations; virologic potencies appear comparable. Since the last edition of the guidelines, data continue to support use of lamivudine or emtricitabine (with these drugs considered interchangeable) as one of the dual nRTI components because each of these thiacytidine compounds is well-tolerated and potent. Lamivudine is available in fixed-dose combinations with either zidovudine or abacavir and emtricitabine has been coformulated with tenofovir disoproxil fumarate. As dual nRTI components, zidovudine and lamivudine, abacavir and lamivudine, and tenofovir and emtricitabine all produce 1.5 to 2.0 log10 copies/mL reductions in plasma HIV-1 RNA levels and early virologic failure usually selects for the M184V substitution associated with resistance to lamivudine or emtricitabine. Thus, assuming the presence of fully drug-susceptible virus, the choice of the dual nRTI component relates to the toxicity profiles and predicted tolerance of zidovudine, abacavir, or tenofovir. Differentiating adverse effects include headache, nausea, anemia, and lipoatrophy for zidovudine; hypersensitivity reaction with abacavir; and renal dysfunction in patients with baseline renal compromise with tenofovir.35 As a fixed-dose combination, zidovudine and lamivudine is given twice daily; abacavir and lamivudine and tenofovir and emtricitabine are given once daily.
In a recent randomized controlled trial, zidovudine plus lamivudine was compared with tenofovir plus emtricitabine, each in combination with efavirenz in 517 treatment-naive participants.35 Tenofovir and emtricitabine with efavirenz resulted in 80% plasma viral suppression below 50 HIV-1 RNA copies/mL at 48 weeks compared with 70% for zidovudine and lamivudine. The former was also superior in CD4 cell count responses and adverse events. There were more discontinuations in the zidovudine and lamivudine group. Better tolerance of tenofovir and emtricitabine, rather than differences in intrinsic antiretroviral activity, may explain these results.
Important data for the field concerning an nRTI-sparing strategy are pending from the analysis of ACTG A5142, which is comparing lopinavir and ritonavir plus 2 nRTIs; efavirenz plus 2 nRTIs; and lopinavir and ritonavir plus efavirenz.
Recommendations
Either of 2 basic 3-drug regimens continues to be recommended for initial therapy: NNRTI-based or PI ritonavir-boosted-based combinations. Of the NNRTIs, efavirenz is recommended due to its consistent efficacy demonstrated in numerous randomized trials and its toxicity profile (AIa).35-37 Efavirenz is not recommended for women in the first trimester of pregnancy (AIIa). Nevirapine is recommended as the NNRTI component for women in whom pregnancy may occur on treatment or who are pregnant and have fewer than 250 CD4 cells/μL (AIIa). Nevirapine is also recommended as an alternative NNRTI in men or women in whom the central nervous system toxicity of efavirenz is not tolerated or does not abate within 2 to 3 weeks of starting treatment.38 The drug should be avoided as initial therapy in women with CD4 cell counts higher than 250/μL and in men with CD4 cell counts higher than 400/μL (AII).
Of the ritonavir-boosted PIs, recommended components are lopinavir (AIa), atazanavir (BIII), fosamprenavir (BIII), or saquinavir (BIII). More data exist for lopinavir and ritonavir but the hyperlipidemia and other metabolic consequences of therapy also support use of atazanavir and ritonavir. Induction of hyperlipidemia with atazanavir and ritonavir is lower than lopinavir and ritonavir; the most frequent agent-specific adverse effect of atazanavir is asymptomatic hyperbilirubinemia. Data comparing lopinavir and ritonavir and atazanavir vs ritonavir in treatment-naive persons are not yet available and will be important. The efficacy of the soft-gel formulation of saquinavir and ritonavir was inferior to lopinavir and ritonavir33 but this was probably due to differences in tolerability and the hard-gel formulation has good virologic efficacy. Data on fosamprenavir and ritonavir support its use in initial regimens. The choice is dependent on provider and patient preference.
Recommended nRTI components in the initial regimen are tenofovir and emtricitabine (AIa), zidovudine and lamivudine (AIa), or abacavir and lamivudine (AIa). Tenofovir is well tolerated but should be used with caution, or avoided, in patients with preexisting renal insufficiency (AIa).
There are almost 20 years of accrued data for zidovudine but, as a thymidine analogue, it produces adverse gastrointestinal tract, central nervous system, and mitochondrial effects more frequently than tenofovir or abacavir. Abacavir, in combination with lamivudine, has comparable antiretroviral activity with the other dual nRTI components listed. However, abacavir-containing regimens carry a 5% to 8% risk of discontinuation due to a hypersensitivity reaction. The risk of abacavir hypersensitivity has been associated with the HLA-B-5701 genotype in some populations and genotypic profiling of patients for whom abacavir therapy is being considered.39-40 Furthermore, abacavir retains activity against viruses with the M184V substitution that occurs commonly with regimens containing lamivudine or emtricitabine, making the drug useful in constructing regimens in which these nRTIs have failed. Table 2 presents the considerations for each nRTI backbone.
Other nRTIs can be combined for initial regimens if none of the recommended combinations can be used. However, certain nRTI components should not be combined. Zidovudine and stavudine should not be used because of antagonism (AIa). Stavudine and didanosine should not be used (AIa) because of overlapping toxic effects, and tenofovir and didanosine should not be used in treatment-naive patients with wild type virus (AIa) because of dampened CD4 cell responses and toxic effects.49-52 Its cautious use in treatment-experienced patients can be considered, however, when treatment options are more limited. Also, abacavir and tenofovir should not be used as the dual nRTI component of an initial regimen because of genetic fragility (eg, K65R substitution emergence and impact).
Triple nRTI regimens are inferior to NNRTI- or ritonavir-boosted PI-based regimens and should only be used in highly selected circumstances, such as high risk of toxic effects, drug-drug interactions, or patient nonadherence to an NNRTI or PI and ritonavir component. Of the triple nRTI regimens, the largest experience is with zidovudine, lamivudine, and abacavir but it is inferior to efavirenz-based regimens.26 Zidovudine, lamivudine, and tenofovir is currently under investigation in the Development of Antiretroviral Therapy in Africa trial53 and quadruple nRTI regimens (eg, zidovudine, lamivudine, abacavir, and tenofovir) remain experimental.54
Hepatitis B Virus Coinfection
Management of persons coinfected with HBV is complicated by 2 factors. First, several of the agents (tenofovir, lamivudine, emtricitabine) used to treat HIV are also active against HBV. Second, as HBV-specific immunity is reconstituted with successful antiretroviral therapy, severe flares of hepatocellular inflammation can occur. These flares may be particularly severe if therapy with 1 or more agents active against HBV is stopped after a period of antiretroviral therapy during which HBV-specific immunity has been restored. Flares have also been observed when HBV resistance to lamivudine or emtricitabine develops while the patient is taking antiretroviral therapy.105-107
Recommendations
When it is necessary to treat both HBV and HIV in coinfected patients, tenofovir and emtricitabine (or lamivudine) are the recommended nRTIs (BIII).108-109 If a HAART regimen is started that includes lamivudine or emtricitabine but not tenofovir, addition of entecavir should be considered to avoid exposing HBV to monotherapy with either lamivudine or emtricitabine (BIII).110
For patients with early HIV infection for which therapy is not yet indicated but who need treatment solely for their HBV infection, adefovir or entecavir may be used with a low risk of selecting for HIV resistance mutations (BIII).111 Entecavir appears to be more potent against HBV than adefovir, but controlled trials have not been reported in the coinfected population (BIII).
Hepatitis C Virus Coinfection
Hepatitis C virus infection complicates the treatment of HIV infection primarily because the underlying liver disease may result in more uncertainty about whether elevations in liver enzymes are due to the HCV or to the antiretroviral regimen. Despite this, there is no evidence that antiretroviral drugs are inherently more hepatotoxic in this population or that there should be a different set of considerations about which agents to use, except for patients who are receiving simultaneous ribavirin treatment of HCV.112
Recommendations
Selection of antiretroviral agents in the HCV-coinfected population should be made with the same considerations as those used in the HCV-uninfected population (BIII). Didanosine should be avoided in patients receiving ribavirin because of an increased risk of pancreatitis and lactic acidosis with this combination (AIIa).113
Race and Sex Differences
Compared with men, women have been shown to have differences in HIV viral load,114-115 drug-related toxic effects,116-121 and pharmacokinetics.122-126 However, there are few data to guide decision making on choice of therapy and dosing by sex. It is increasingly important that clinical trials be designed to address issues important for the care of women with HIV and that women are enrolled in larger numbers in clinical trials.
There are no data that indicate racially based differences in the responsiveness to antiretroviral therapy.127 A genetic polymorphism found in 20% of African Americans reduces the metabolism of efavirenz, thereby leading to higher average drug levels.128 Although this polymorphism is associated with more adverse central nervous system events in patients taking efavirenz, its frequency in the African American population does not result in a higher rate of adverse effects necessitating drug discontinuation.128 Conversely, the HLA-B 5701 haplotype associated with abacavir hypersensitivity in the white population is less common in the black population.39 It is likely that other such polymorphisms will be delineated in the future, but at this point they do not dictate changes in the approach to antiretroviral chemotherapy.
Recommendation
Antiretroviral therapy decisions should be made independent of the race of the patient (BIIa).
Pregnancy
The dual goals of antiretroviral therapy in pregnant women are to provide therapy for the mother and to reduce the likelihood of transmission of the virus to the fetus or neonate. Indications for therapy in pregnant women generally mirror those in other HIV-1-infected adult populations but the choices of antiretroviral agents are more limited because of concerns regarding potential teratogenicity.
Recommendations
The initiation of antiretroviral therapy during the first trimester should be avoided if possible. In general, when an HIV-1 infected woman taking effective antiretroviral therapy becomes pregnant, antiretroviral drugs should not be discontinued although an adjustment in the regimen based on the considerations outlined below may be in order. After the first trimester of pregnancy, the indications for the initiation of therapy are the same as in nonpregnant women except that therapy directed at preventing viral transmission to the fetus is generally recommended during the third trimester for all women regardless of the CD4 cell count. If antiretroviral drugs are administered to women to prevent maternofetal transmission of HIV-1, they should be given in combinations intended to be fully suppressive.
Assuming the virus is susceptible, zidovudine and lamivudine or emtricitabine are the preferred nRTIs.99 Other nRTIs may be substituted if resistance testing indicates that drug resistance mutations are present (BIII).
An increased risk of hepatotoxicity is associated with the use of nevirapine in pregnancy, especially if initiated in women with more than 250 CD4 cells/μL.100 In women who become pregnant while taking nevirapine, this risk is substantially lower and although close monitoring is warranted, it is not required that nevirapine be replaced in women who are receiving it without untoward effects when they become pregnant (BIII).
Until more data are available that address concerns about bone formation in utero, tenofovir should be avoided unless resistance testing suggests its use is advisable (BIII). Efavirenz is contraindicated in the first trimester of pregnancy (AII).101-103 Nelfinavir has been used extensively in pregnancy, but concerns about its potency make it a less attractive agent than in the past (BIIa).104
|
| |
|
|
|
|
|