 |
|
|
| |
CCR5 Antagonists: When Should They be Used?
|
| |
| |
A report from the XVI International AIDS Conference
Toronto, August 13-18, 2006
Mark Mascolini
For all their promise and proximity to licensing, CCR5 antagonists remain among the most problematic antiretrovirals yet advanced for treatment of HIV infection. Concerns over this new class fall into three bins--coreceptor tropism, side effects, and optimal time of use:
Coreceptor tropism is the fancy term for which coreceptor on T cells HIV prefers. After it snags the CD4 receptor itself, HIV must also grasp one of two coreceptors, CCR5 or CXCR4. R5-tropic virus favors CCR5, and X4-tropic HIV favors CXCR4. Dual-tropic means an individual virus can use either CCR5 or CXCR4; mixed-tropic means the viral population contains a mix of R5- and X4-tropic virus.
As soon as researchers discovered the coreceptors, others began fashioning drugs that might keep HIV from snaring them. But even before candidate compounds emerged, a theoretical worry arose: Would shielding CCR5 receptors from HIV drive a person's viral swarm to target CXCR4 instead?
The answer to that question is terribly important for two reasons: First, CCR5 antagonists do nothing to stop replication of purely X4-tropic virus. And second, X4-tropic HIV--if it does start to dominate a person's viral horde--signals advanced disease. If X4-tropic virus contributes to HIV disease progression, keeping it at bay would be a great idea. Three studies at the XVI International AIDS Conference addressed emergence of X4-favoring virus and two of them addressed CCR5 antagonist activity against viral pools aswim with X4 virus.
Side effects have already sunk one CCR5 antagonist, the Glaxo candidate, aplaviroc. But the most frightening potential side effects seen so far--liver failure and cancer--seem either limited to one CCR5 plugger or (in the case of cancer) still impossible to blame on these drugs. Severe liver toxicity stopped aplaviroc's licensing sprint cold, but so far liver troubles seem rare with Pfizer's maraviroc or Schering's vicriviroc. AIDS Conference attendees heard more details of the malignancies that arose in people taking vicriviroc [1], but cancer has not cropped up in people taking maraviroc [2]. Indeed, maraviroc's side effect profile looks pretty clean so far.
When to use CCR5 antagonists--early in the course of infection (when CCR5-using virus holds sway), or later (when people with multidrug resistance desperately need new drugs)--will certainly depend on a host of individual patient factors. But some experts fear the focus on CCR5 drugs as salvage options may unjustly overshadow their prime value earlier in the disease course. The CCR5 studies unveiled in Toronto and other recent research address this pivotal question.
Vicriviroc activity against X4-R5 mixes
Two talks on clinical trials of vicriviroc and maraviroc at Toronto's brimful late-breaker session came laden with potential implications on how CCR5 antagonists may alter the course of HIV infection. Indeed, these two intriguing trials may say as much about coreceptor conundrums as about the two drugs themselves. The maraviroc study did so by sizing up this agent in treatment-experienced people with some X4-favoring virus when the study started. The vicriviroc trial also involved people with plenty of treatment experience, and they all signed up carrying nothing but R5-tropic virus. But that changed.
Roy Gulick (Cornell University, New York) and ACTG A5211 colleagues randomized 118 treatment-experienced people to placebo or to 5, 10, or 15 mg of vicriviroc daily for 14 days [1]. At that point everyone added a so-called optimized background regimen (OBR) of other antiretrovirals picked on the basis of resistance tests and drug history, and follow-up continued to week 24. Everyone who enrolled had to be taking a ritonavir-containing regimen, and everyone had to have triple-class experience; one third had already tried the fusion inhibitor enfuvirtide. Median starting viral load measured 36,380 copies and median CD4 count 146.
The study protocol stipulated that only people with R5-tropic virus could sign up. And accordingly no volunteers had X4 virus when screened for the study with an assay that rates HIV for R5 or X4 leanings. But when people actually started their study assignment--the baseline point--10% had dual- or mixed-tropic virus. Does this mean viral populations in these 13 people started shifting coreceptor allegiance in just a few weeks' time in all these people? Not necessarily. As Mike Youle observes in his review of this trial for NATAP (see note 3), that slippage in exclusive R5 tropism underlines the current assay's imperfections.
Furthermore, in people with no evidence of X4 virus when treatment began, the vicriviroc dose assigned appeared to affect not only viral load and CD4 response, but also further shifts in coreceptor preference (Table 1). Only 1 person in the placebo group (4%) had a shift to dual or mixed tropism, compared with 2 (7%) in the 15-mg arm, 3 (10%) in the 10-mg arm, and 8 (27%) in the 5-mg arm. Compared with 60 people taking 10 or 15 mg of vicriviroc daily, the 30 taking 5 mg of vicriviroc daily had marginally lower viral load drops at day 14 and week 24 and much smaller CD4 gains at week 24. None of the differences between vicriviroc arms reached statistical significance in this small study, but the ACTG shut down the 5-mg arm, partly because a low dose also performed particularly poorly in a separate trial involving previously untreated people [4].
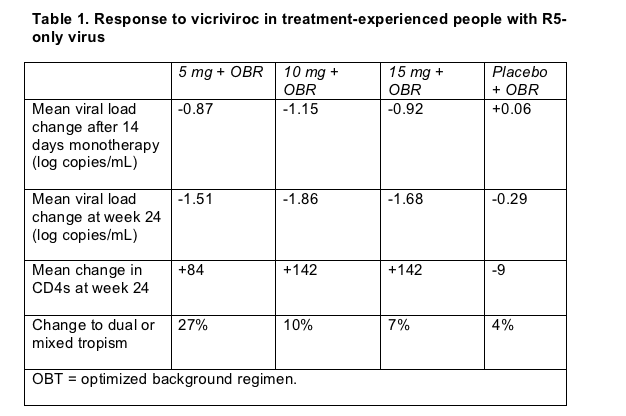
Limiting the analyses to people taking 10 or 15 mg of vicriviroc daily, Gulick measured a 1.83-log viral load drop at week 24 in 71 people with no evidence of X4 tropism at baseline versus a 0.77-log ebb in 10 people with dual or mixed tropism at baseline, a significant difference (P = 0.01). That difference comes as no surprise since vicriviroc has no activity against X4-favoring virus. The greatly diminished but still-decent 0.77-log drop in people with X4-R5 mixes reflects the drug's activity against the R5 part of the viral flock, coupled with the background regimen's activity against various parts of the viral population.
Sixteen of 30 people (53%) in the 10-mg arm, 14 of 30 (47%) in the 15-mg arm, and 3 of 28 (11%) in the placebo group reached a viral load below 400 copies by week 24. Twelve (40%) taking 10 mg, 8 (27%) taking 15 mg, and 2 (7%) taking placebo got their load under 50 copies at that point.
Do CCR5 antagonists promote X4 virus?
Variability in the coreceptor assay almost certainly explains some of the drift toward X4 tropism in the vicriviroc trial. How much vicriviroc itself (or maraviroc) contributes to this drift is the million-dollar question--perhaps literally for CCR5 antagonist marketers. I asked two coreceptor experts (John Moore from Cornell University and Robert Doms from the University of Pennsylvania) and one HIV clinician who studies this issue (Steven Deeks from the University of California) for their opinion on whether CCR5 antagonists promote emergence of dual- or mixed-tropic viruses. All said perhaps, but the evidence to date has bowled over none of them.
"I believe the risks of this happening are real," Moore replied by e-mail, "but have been overblown to date, partly as a result of inadequacies in the tropism detection assays." In lab studies, he points out, the path HIV takes to resistance against CCR5 drugs does not involve a jump to the CXCR4 coreceptor. The resistance pathway may prove different in humans, "but as of today there's no evidence of that [jump] happening." Other mechanisms may also foster a hop to X4 during CCR5 antagonist therapy, Moore adds, but those routes remain theoretical.
Deeks spells out a critical distinction when weighing how CCR5 agents may affect viral tropism. "The evidence to date suggests that R5 inhibitors will selectively expand pre-existing X4 variants," he writes. "I am unaware of any data suggesting that R5 inhibitors promote the phenotypic evolution of an R5 virus to an X4 variant."
Doms thinks the evidence that CCR5 drugs will open the door to X4-tropic virus is "not compelling--basically we don't know." In fact, pointing to the "inherent variability" in both HIV and the infected person, he doesn't believe a single pathway to CCR5 antagonist resistance will emerge. Although Doms states his "bias" that "some X4 viruses will emerge upon R5 inhibitor therapy . . . in reality we are all guessing." He does think, though, that clinical trials will provide the answers.
Cancer cases in people taking vicriviroc
After 24 weeks rates of grade 3 or 4 lab or clinical problems did not differ significantly between groups in the vicriviroc trial. Malignancies developed in 7 people--5 taking vicriviroc, 1 taking placebo, and one taking placebo for 7 months then vicriviroc for 3 months. In the last person localized perianal squamous cell carcinoma appeared 1 month after this volunteer stopped taking vicriviroc. Among the 5 people who took only vicriviroc during the trial, non-Hodgkin lymphoma arose in 2 people, 1 taking 5 mg and 1 taking 15 mg; 1 of these 2 had Hodgkin disease earlier. Clinicians diagnosed Hodgkin disease in 1 person taking 5 mg of vicriviroc and 1 taking 10 mg, and 1 of these 2 had non-Hodgkin lymphoma earlier. Gastric adenocarcinoma developed in 1 person taking 15 mg of vicriviroc.
No one knows if this rash of cancers in a 24-week trial reflects cruel coincidence or something more ominous. Researchers studying maraviroc in 109 treatment-experienced people (see below [2]) saw no cancers through 24 weeks of follow-up. And Glaxo reported no malignancies among people taking the CCR5 plugger they withdrew because of liver toxicity.
A few groups have advanced theories to explain why blocking CCR5 may damage the liver [5-7]. But these theories do not explain why--if this is a CCR5 antagonist class effect--only one case of bad liver trouble has emerged in a person taking maraviroc, and that person had hepatitis C virus coinfection and took other hepatotoxic drugs. In the maraviroc trial presented in Toronto, ominous liver enzyme readings proved no more likely with the CCR5 antagonist than with placebo in people with plenty of antiretroviral experience and advanced HIV infection [2].
A causal link between blocking CCR5 and cancer seems even more tenuous. Indeed, most cell studies published so far suggest that CCR5 antagonists could prevent or delay cancer because CCR5 studs some tumor cells [8-11]. Also, HIV-infected people with partial innate suppression of CCR5 expression (that is, people carrying one mutant CCR5-delta32 gene and one nonmutant gene) appear to have a lower-than-expected rate of malignant non-Hodgkin lymphoma [12] On the other hand, one study of 547 women not tested for HIV did find that the CCR5-delta32 mutation shortens disease-free survival in women with wild-type p53 gene tumors [13]. But breast cancer has not been tied to CCR5 antagonist therapy.
People who carry matching CCR5-delta32 genes (homozygotes) cannot be infected with CCR5-tropic HIV because the delta32 deletion prevents CCR5 expression on CD4 cells. Yet these people, about 1% of white Europeans, apparently have normal life spans. But noted HIV immunologists say this fact offers no assurance that blocking CCR5 with antagonists will prove similarly benign. "While compensation for the congenital absence of CCR5 by a redundant network of chemokine-ligand interactions may be reasonably well tolerated unless severely challenged," write Michael Lederman and colleagues, "it is not clear whether acute effects of pharmacological or immunologic CCR5 blockade will be as well tolerated" [14].
Maraviroc in people with X4-R5 blends
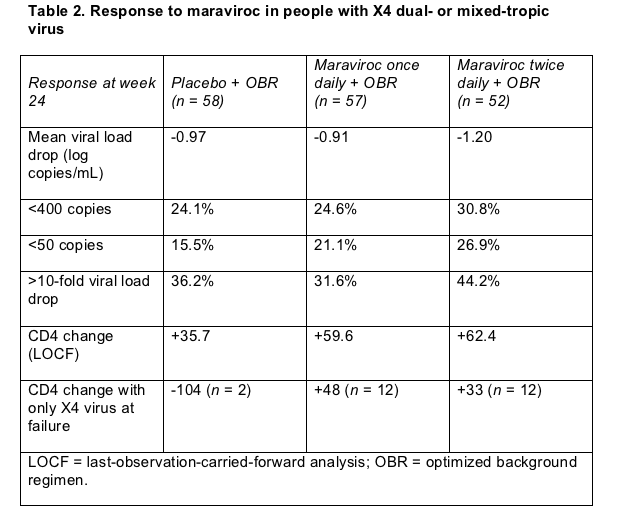
Toronto's maraviroc late-breaker yielded an even bigger horde of data addressing tropism worries because the trial set out to study this drug's prowess in people with dual- or mixed-tropic HIV pools [2]. Compared with placebo, maraviroc did little or nothing to stymie HIV in this trial, but it did not hasten disease progression, as one might fear if X4 virus starts to gain the upper hand. In fact, CD4 counts rose more with 24 weeks of maraviroc than with placebo.
Pfizer's Howard Mayer and an international cast of collaborators recruited people with a viral load at or above 5000 copies and either triple-class experience or resistance to two antiretroviral classes. Among people with at least some X4 virus at screening, 58 got randomized to placebo plus a background regimen, 57 to 150 mg of maraviroc once daily plus other drugs, and 52 to 150 mg of maraviroc twice daily plus other drugs. These people had advanced HIV infection reflected in average CD4 counts below 100 and average viral loads above 100,000 copies in all treatment arms. More than half in each group included enfuvirtide in their regimen, and about two thirds in each group took two to four active drugs besides maraviroc.
Twice-daily maraviroc did slightly (but not significantly) better than once-daily maraviroc or placebo in lowering viral loads through 24 weeks, the trial's primary endpoint (Table 2). Marginally more people taking the twice-a-day dose reached viral loads below 400 and 50 copies at week 24, but again these differences from the placebo arm did not reach statistical significance.
People taking either dose of maraviroc did gain more CD4 cells than people taking placebo (Table 2). And among the handful of people who had only X4-tropic virus at virologic failure, this CD4 benefit with maraviroc looked striking when compared with placebo.
Do these findings mean CCR5 antagonists--far from speeding disease progression in people with some X4 virus--may actually slow progression? As our expert troika of Moore, Doms, and Deeks explained (above), no one knows for sure whether using CCR5 antagonists will foster ascendance of X4 virus. Research from the early 1990s linked X4 virus to advanced HIV infection [15,16], but whether X4-tropic strains cause disease progression or merely flag its imminent arrival remains open to debate.
"Although the emergence of X4 virus predates and predicts subsequent disease progression," Steven Deeks explained via e-mail, "this observation does not conclusively address the cause vs. effect question. Strong evidence for a causal role comes from the anecdotal experience that infection with X4/dual-tropic virus is associated with rapid disease progression" [see note 17], but, he adds, the just-discussed maraviroc results did not confirm that suspicion. "If anything, there was evidence of immunologic benefit" with maraviroc. Deeks' own clinical data (below) suggest that in people taking partially suppressive antiretroviral therapy, "the presence of X4/dual tropic virus is not associated with an appreciable increase in the rates at which CD4 T cells are lost."
John Moore sounds less optimistic. "I believe X4 viruses contribute to progression," he writes, "and that the sustained emergence of X4 viruses is a problem, clinically. The key question, however, is whether the kind of X4 assay signals being generated in the CCR5 inhibitor trials, at baseline or after therapy, are meaningful. There's a difference between a low-level, transient X4 'blip' that registers as positive in the assay, and a more sustained, higher-level signal of the type that's been associated with more rapid disease progression over the years. I don't get bent out of shape over 'blips'; I would over something more serious. This is exactly the kind of issue we need the trials to sort out--what's a problem signal, and what's a meaningless 'blip'? Right now, we just don't know, and that's the problem."
In the maraviroc trial virologic failure with the CCR5 antagonist did correlate with emergence of an X4-heavy viral swarm. When the background regimen plus placebo failed virologically, only 2 people in that arm had X4-only virus. But when maraviroc plus other drugs failed, 12 people in each maraviroc arm had X4-only virus upon failure. As Table 2 shows, though, the 24 people in whom maraviroc fizzled with X4 virus tended to have rising CD4 counts, while the 2 people in the placebo arm had big CD4 deficits at failure. Still, when one considers the low pretrial CD4 counts in this study, it's hard to say how much benefit people derive from 40 or 50 extra CD4 cells at virologic failure with an X4-rife viral population.
Best time to use CCR5 antagonists
Because people with multidrug-resistant virus urgently need antiretrovirals that work differently from protease and reverse transcriptase inhibitors, CCR5 antagonists could fill a yawning salvage niche. But more than one antiretroviral sage maintains that advanced HIV infection may not be the best time to use these novel drugs, for reasons well rehearsed in this article: R5-tropic virus dominates in the early years of HIV infection; X4-tropic virus, when it does appear, appears late.
But a big cohort study presented in Toronto suggested that X4s sometimes make a surprisingly early debut [18]. This analysis of the Multicenter AIDS Cohort Study (MACS) saw X4s in circulation at a median CD4 count when most clinicians would not consider starting antiretroviral therapy--484 cells. Four men in this cohort had X4 virus within 2 years of HIV infection, and all 4 had AIDS or fewer than 200 CD4s within 3.5 years.
MACS enrolls gay men at risk for HIV infection and tracks them regularly whether or not they pick up HIV. This analysis sorted 685 blood samples from 67 men who did get infected, rating them for R5 and X4 tropism. The men sampled had a median age of 32 years when infected with HIV, usually in the 1980s.
Two men had mixed R5/X4 virus at their first visit after contracting HIV infection, and their virus retained the knack for yoking either coreceptor throughout follow-up. The other 65 men carried no detectable X4s on their first visit after infection, but 33 (51%) later had R5/X4-using virus.
Half of the men in whom X4 use arose had R5/X4 virus before an AIDS diagnosis or before their CD4 count dropped under 200. Median time from X4 emergence to AIDS or fewer than 200 CD4s measured a mere 1 year. Median CD4 count when assays first picked up X4 stood at 484. The interquartile range for X4 emergence--299 to 718 CD4s--meant most men had more than 300 CD4s when X4-using virus first popped up.
Earlier MACS work defined a point when the T-cell cache in HIV-infected men shifts from relative stability to a progressively downhill drop [19]. The new analysis figured how this so-called inflection point correlates with the change in R5 and X4 use. X4-bent virus appeared in 54% of men with a defined inflection point compared with 24% of those with no defined inflection, a significant difference (P = 0.048). Among men with a defined inflection point who later had X4-using virus, the X4s arrived a median of 9 months after they passed the inflection point.
HIV infection did progress to AIDS in some men who never had evidence of X4 virus, a finding confirming earlier research. But the MACS study challenges the assumption that X4 virus appears only in people with advanced HIV infection.
Around the time of the AIDS Conference, Steven Deeks and Peter Hunt in San Francisco joined colleagues in Vancouver to publish a study comparing coreceptor leanings in antiretroviral-experienced people and still-untreated people [20]. Seventy-five of 182 treated people in the San Francisco and Vancouver cohorts (41%) had dual- or mixed-tropic virus, compared with 178 of 976 treatment-naive people (18%), an unsurprisingly significant difference (P < 0.001) that remained significant after statistical tweaking for CD4 count and CCR5-delta32 genotype. Even so, the advent of X4 virus in nearly 1 in 5 untreated people confirms that double coreceptor use can appear relatively early in the course of infection.
Treated people with a detectable viral load proved four times more likely to have dual- or mixed-tropic HIV than treatment-naive people, regardless of antiretroviral regimen, CD4 count, or CCR5-delta32 genotype. Because lower CD4 nadirs explained most of the X4 population, Deeks and coworkers observe that "X4-tropic variants apparently persist despite treatment-mediated restoration of peripheral CD4+ T cell counts." And since treated people with drug resistance and detectable viremia apparently have a slimmer chance of suppressing HIV with a CCR5 antagonist regimen, they suggest that "CCR5 inhibitors may thus be best strategically used before salvage therapy and before significant CD4+ T cell depletion."
In their e-mail comments, Deeks, Doms, and Moore all agree that--given what we know about when X4 virus appears--CCR5 antagonist therapy makes most sense as an early strategy. But certainly no HIV clinician will forego using these drugs in people with advanced disease, even if they suspect--or know--that X4s abound in their lymphoid tissue. Still, should people with advanced disease and no detectable R5 virus take drugs with a still-murky side effect profile and no activity against X4 virus?
Most clinicians can probably answer that question right now, but the only way they can tell how much R5 versus X4 a person carries is to assay their virus for coreceptor cravings. Though our three experts--and anyone else you ask--will tell you that current assays have distinct limits, Deeks, Doms, and Moore all see a role for coreceptor testing.
Moore writes that clinicians will probably have to use these assays before prescribing a CCR5 antagonist, but "there are real concerns about how good these assays are on a quantitative level, with minor blips in X4 being interpreted as clinically meaningful when they are unlikely to be."
Doms concurs that coreceptor typing "would be a wise thing to do until such time that we as a field determine that phenotype measures are no longer necessary, or are no longer necessary in some cases (patients with high CD4, for example)."
Deeks notes that, as the maraviroc study shows, "the use of R5 inhibitors may not cause harm in patients with dual-tropic virus," though the virologic benefit looked limited. "To more carefully define the level of potential benefit versus risk," he suggests, "some coreceptor tropism assays will almost always be preferable."
For clinicians who do ponder prescribing CCR5 thwarters earlier in HIV's course, one mammoth if remains: CCR5 antagonists make more sense for early infection if they work as well as other antiretrovirals early on. And as everyone who follows CCR5 studies knows, vicriviroc did not stop HIV as well as efavirenz in a randomized trial involving treatment-naive people [4]. But it's tough to interpret results of this prematurely closed trial because its controversial design included a 2-week run-in with vicriviroc monotherapy.
Some observers feared that vicriviroc-resistant virus gained a toehold in those 2 weeks of monotherapy and contributed to the drug's later failure. At the 2006 Resistance Workshop Schering offered data they read as evidence against emergence of resistant virus in this trial [21]. But two resistance mavens analyzing this study saw things differently.
"I do not think these data constitute evidence that resistance was selected during that initial 2-week monotherapy lead-in," said Harvard's Daniel Kuritzkes, "but it is clear to me that many of the patients who were failing vicriviroc did fail with vicriviroc resistance" [22].
"Right now the tools we are using to try to understand reduced response to CCR5 antagonists are inadequate," added Jonathan Schapiro from Tel Aviv's Sheba Medical Center. "The fact that we do not see classical resistance emergence of the sort we are used to detecting with other antiretrovirals does not necessarily mean the virus in these cases is not becoming insensitive to CCR5 antagonists."
If HIV did become resistant to vicriviroc in this trial, as Kuritzkes and Schapiro maintain, when it became resistant matters. Emergence of resistance during 2 weeks of monotherapy would be disappointing, though not hugely surprising. But if vicriviroc-resistant virus evolved when people were taking the CCR5 antagonist with Combivir (AZT/3TC), as they did after the monotherapy phase, it would suggest vicriviroc is more prone to resistance than efavirenz, the alternative drug in this randomized comparison.
With colleagues in Europe and Canada, Schering randomized 92 treatment-naive people to 2 weeks of placebo or 25, 50, or 75 mg of vicriviroc once daily [4]. After 2 weeks everyone added Combivir and the placebo group swapped their dummy pills for efavirenz. A review panel shut down the trial when they saw significantly more virologic breakthroughs in the vicriviroc arm. Whereas 4% of people taking efavirenz had breakthroughs above 50 copies, breakthroughs hit 56% in people taking 25 mg of the CCR5 antagonist (P < 0.001 versus efavirenz), 41% in those taking 50 mg (P = 0.003), and 17% in those taking 75 mg (a nonsignificant difference versus efavirenz, P = 0.183).
In an earlier phase 2 trial of vicriviroc monotherapy in naive or currently untreated people [23], twice-daily doses of 10 mg, 25 mg, and 50 mg pushed viral loads down farther than the once-daily doses in the more recent study. A new phase 2 trial involving treatment-experienced people will assess 20 and 30 mg of vicriviroc daily plus a ritonavir boost.
Whether CCR5 antagonists will make the grade as first-line antiretrovirals depends on both their potency and safety. As Steven Deeks observes in a recent Lancet essay, today's favored first-line combos have enviable potency and safety records [24]. He reasons that displacing agents from these regimens will take some doing.
So far CCR5 antagonists have not proved themselves equal to first-line mainstays in previously untreated people, though results of a phase 3 trial comparing maraviroc (300 mg twice daily) with efavirenz in antiretroviral-naive people remain to be reported. (A safety panel closed the 300-mg once-daily maraviroc arm.) At the same time, glaring questions on CCR5 antagonist safety still need answers. Even if current CCR5 candidates pose little threat of liver toxicity, and even if the malignancies seen with vicriviroc happen to be happenstance, these drugs may have other unkind consequences. Michael Lederman, Donald Mosier, and colleagues suggest two possibilities--a blunted response to certain vaccines and impaired revival of healthy gut lymphoid tissue [14].
Mark Mascolini writes about HIV infection (markmascolini@earthlink.net).
References and Notes
1. Gulick R, Su Z, Flexner C, et al. ACTG 5211: phase II study of the safety and efficacy of vicriviroc in HIV-infected treatment-experienced subjects. XVI International AIDS Conference. August 13-18, 2006. Toronto. Abstract THLB0217.
2. Mayer H, van der Ryst E, Saag M, et al. Safety and efficacy of maraviroc, a novel CCR5 antagonist, when used in combination with optimized background therapy for the treatment of antiretroviral-experiences subjects infected with dual/mixed-tropic HIV-1: 24-week results of a phase 2b exploratory trial. XVI International AIDS Conference. August 13-18, 2006. Toronto. Abstract THLB0215.
3. Mike Youle from London's Royal Free Hospital discusses CCR5 antagonists and other investigational antiretrovirals in an article on the NATAP site (http://www.natap.org/2006/IAS/IAS_91.htm).
4. Greaves W, Landovitz R, Fatkenheuer G, et al. Late virologic breakthrough in treatment-naive patients on a regimen of Combivir + vicriviroc. 13th Conference on Retroviruses and Opportunistic Infections. February 5-8, 2006. Denver. Abstract 161LB.
5. Moreno C, Gusto T, Nicaise C, et al. CCR5 deficiency exacerbates T-cell-mediated hepatitis in mice. Hepatology 2005;42:854-862.
6. Mauss S, Puoti M. CCR5 deficiency exacerbates T-cell-mediated hepatitis in mice--clinical implications for CCR5 inhibition as antiretroviral therapy. 2006;43:879; author reply 879-880.
7. Ajuebor MN, Carey JA, Swain MG. CCR5 in T cell-mediated liver diseases: what's going on? J Immunol 2006;177:2039-2045.
8. Vaday GG, Peehl DM, Kadam PA, Lawrence DM. Expression of CCL5 (RANTES) and CCR5 in prostate cancer. Prostate 2006;66:124-134.
9. Oba Y, Lee JW, Ehrlich LA, et al. MIP-1alpha utilizes both CCR1 and CCR5 to induce osteoclast formation and increase adhesion of myeloma cells to marrow stromal cells. Exp Hematol 2005;33:272-278.
10. Robinson SC, Scott KA, Wilson JL, et al. A chemokine receptor antagonist inhibits experimental breast tumor growth. Cancer Res 2003;63:8360-8365.
11. Uekusa Y, Yu WG, Mukai T, A pivotal role for CC chemokine receptor 5 in T-cell migration to tumor sites induced by interleukin 12 treatment in tumor-bearing mice.
Cancer Res 2002;62:3751-3758.
12. Dean M, Jacobson LP, McFarlane G, et al. Reduced risk of AIDS lymphoma in individuals heterozygous for the CCR5-delta32 mutation. Cancer Res 1999;59:3561-3564.
13. Manes S, Mira E, Colomer R. et al. CCR5 expression influences the progression of human breast cancer in a p53-dependent manner. J Exp Med 2003;198:1381-1389
14. Lederman MM, Penn-Nicholson A, Cho M, Mosier D. Biology of CCR5 and its role in HIV infection and treatment. JAMA 2006;296:815-826.
15. Schuitemaker H, Koot M, Kootstra NA, et al. Biological phenotype of human immunodeficiency virus type 1 clones at different stages of infection: progression of disease is associated with a shift from monocytotropic to T-cell-tropic virus population. J Virol 1992;66:1354-1360.
16. Koot M, Keet IP, Vos AH, et al. Prognostic value of HIV-1 syncytium-inducing phenotype for rate of CD4_cell depletion and progression to AIDS. Ann Intern
Med 1993;118:681-688.
17. At least two case reports indicate rapid progression after infection with multidrug-resistant virus that uses both CXCR4 and CCR5: Markowitz M, Mohri H, Mehandru S, et al. Infection with multidrug resistant, dual-tropic HIV-1 and rapid progression to AIDS: a case report. Lancet 2005;365:1031-1038; Masquelier B, Capdepont S, Neau D, et al. Virological characterization of an infection with a dual-tropic, multidrug resistant HIV-1 leading to rapid immunodeficiency. 4th European HIV Drug Resistance Workshop. March 29-31, 2006. Monte Carlo. Abstract 7.
18. Shepherd J, Jacobson L, Qiao W, et al. Analysis of the timing of CXCR4-tropic HIV emergence in the MACS cohort. XVI International AIDS Conference. August 13-18, 2006. Toronto. Abstract TUPE0001.
19. Gange SJ, Munoz A, Chmiel JS, et al. Identification of inflections in T-cell counts among HIV-1-infected individuals and relationship with progression to clinical AIDS. Proc Natl Acad Sci USA 1998;95:10848-10853.
20. Hunt PW, Harrigan PR, Huang W, et al. Prevalence of CXCR4 tropism among antiretroviral-treated HIV-1-infected patients with detectable viremia. J Infect Dis 2006;194:926-930.
21. Landovitz R, Faetkenhauer G, Hoffmann C, et al. Characterization of susceptibility profiles for the CCR5 antagonist vicriviroc in treatment-naive HIV-infected subjects. XV International HIV Drug Resistance Workshop. June 13-17, 2006. Sitges, Spain. Abstract 18.
22. Kuritzkes DR, Schapiro JM. 2006 International HIV Drug Resistance Workshop. Clinical Care Options. August 2006. (http://www.clinicaloptions.com/HIV/Conference%20Coverage/Resistance%202006.aspx).
23. Schuermann D, Pechardscheck C, Rouzier R, et al. SCH 417690: antiviral activity of a potent new CCR5 receptor antagonist. 3rd International AIDS Society Conference on HIV Pathogenesis and Treatment. July 24-27, 2005. Rio de Janeiro. Abstract TuOa0205.
24. Deeks SG. Challenges of developing R5 inhibitors in antiretroviral naive HIV-infected patients. Lancet 2006;367:711-713.
|
| |
|
|
|
|
|