 |
|
|
| |
New Antiretrovirals
|
| |
| |
Toronto: XVI WAC Aug 2006
Mike Youle, MD, Royal Free Hospital, London
Earlier this summer I gave series of lectures in Canada, one of which was in the same hotel in Vancouver where almost exactly 10 years before we had heard the first seriously good results from protease inhibitor (PI) based HAART. The therapeutic landscape has changed dramatically since then with better tolerated drugs and formulations, fewer pills per regimen and a shift towards once daily therapy. The last patient to die in my cohort of AIDS with no therapy options left was in May 2000 and the major cause of HIV disease and death in the UK is late presentation. This, of course, is not the case for much of the globe and the XVI International AIDS Conference reflected this in the prominence rightly given to access and empowerment issues. It struck me as highly ironic that the activists chose to target Abbott for its pricing issues but completely ignored the systematic genocide by default being committed in South Africa, where the Teflon coated Minister of Health seems to be hell-bent on preventing the use of appropriate therapy and where more infections occur daily than patients enter onto HAART therapy programs. There was however some excellent discussion on this issue by researchers from South Africa.
Now, to get onto my normal topic of what's hot in the therapy pipeline. Although this meeting is not primarily scientific, there was a selection of data for many of the new compounds in development; these are listed in the table below:
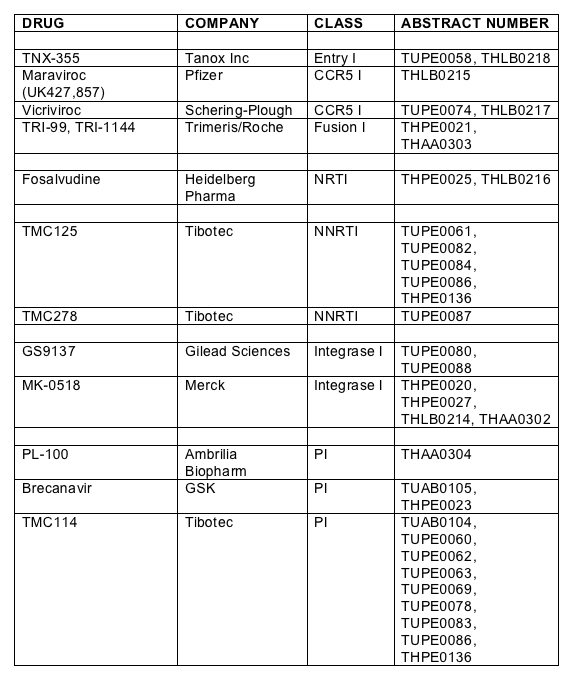
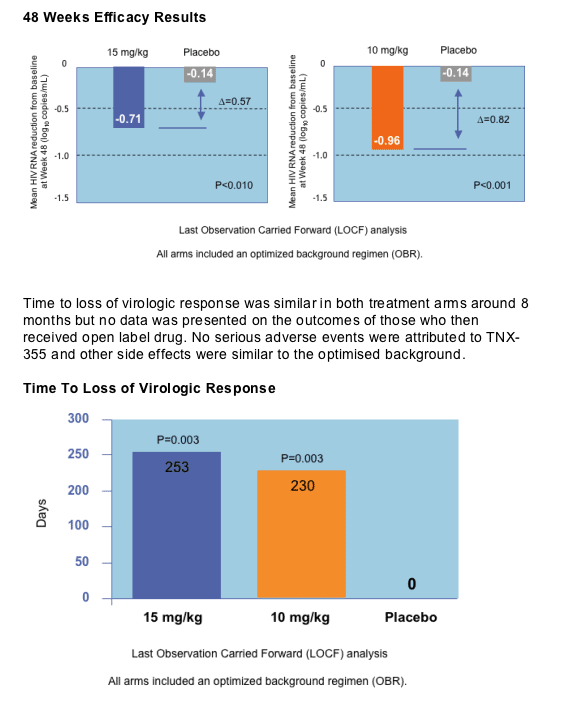
Thomas Duensing from Tanox Inc showed new 48 week data of a randomised double-blind, placebo, and triple-class experienced patient study treated with the novel entry inhibitor TNX-355 which prevents conformational change in gp120 to expose the co-receptor [THLB0218]. TNX-355 is a monoclonal antibody and is given as a weekly intravenous infusion. Subjects were randomised to receive 10mg weekly for 9 doses given thereafter every two weeks, 15mg twice every two weeks or optimized background. Virological failures were rolled over onto TNX-355 plus a new optimised regimen. Eighty two patients with T4>50 and viral load above 10,000 copies were randomised to the three groups. The viral load declines are shown below and are approximately 0.2log less than the equivalent 24 week data, shown previously and data which was on the withdrawn poster [TUPE0058]. Mean increase in T4 was 51 for the 15mg group and 48 for the 10mg group compared to only 1 for the placebo arm (p<0.05).
In a poster further data was shown for the activity of this agent against site directed mutants containing enfurvitide (T20) resistance substitutions (G36D, V38A and N43D) [THPE0024]. HIV envelopes containing these substitutions showed an 11-32 fold reduced sensitivity to T20 but conversely less than two-fold reduced susceptibility to TNX-355 suggesting that cross-resistance may not be a major problem for these two drugs.
In a poster from Angela Sanone from Schering-Plough Research Institute the effect of combining the R5 receptor antagonist, vicriviroc (VCV) and a range of boosted protease inhibitors (atazanavir, saquinavir, indinavir, fosamprenavir and tipranavir; also unboosted nelfinavir) was evaluated in a pharmacokinetic study in healthy volunteers [TUPE0074]. It seems that the addition of any of these agents has negligible effect on the key VCV PK parameters and no significant adverse events were reported. Clearly, further data will be needed with boosted lopinavir and with TMC114 (darunavir).
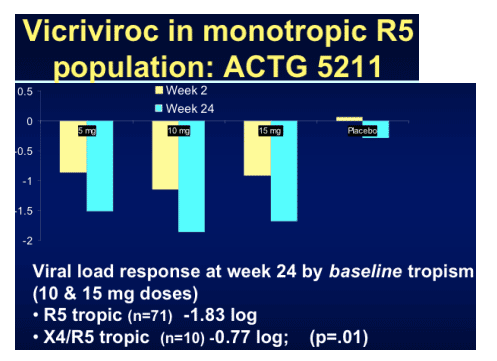
After the disappointment of the earlier vicriviroc studies it was comforting to see some positive news with the late-breaker presentation by Trip Gulick of the ACTG5211 study assessing 3 doses of vicriviroc against placebo in the highly experienced R5-tropic patient population [THLB0217].
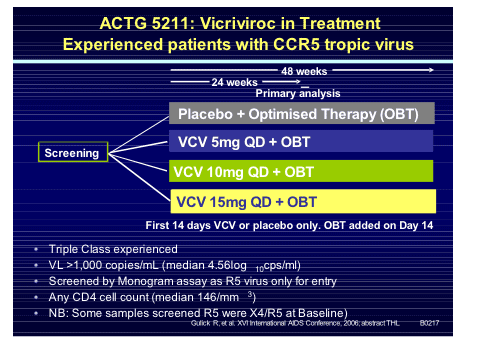
One hundred and eighteen subjects (median VL 36,380 copies/mL and T4 146 cells/mm3) were randomised within the study and viral load declines were significantly greater in all three vicriviroc arms compared to placebo at day 14 and week 24 (p<0.05; ITT analysis). Although not statistically significant the lower dose of 5mg resulted in more virologic failures and greater X4 virus emergence in that arm. Thirty three per cent of participants were already experienced to enfurvitide, 92% male, 66% Caucasian, 20% black and 12% Hispanic. Tropism at study entry was 86% R5 only and 10% dual or mixed tropic. Considering that at screen, a few weeks before this, all were R5 only goes to show some of the limitations of the current technology to exactly decide the tropism of a patients' virus. However, emerging data that even mixed/dual tropic virus responds to R5 receptor blockers may make the use of such an assay less vital.
In the table immediately below, mean change in HIV-1 RNA (log10 c/ml) at Day 14 was -0.87, -1.15, and -0.92 for the VCV 5mg QD, 10mg QD, and 15mg QD (+ OBT) dose groups, compared to -0.08 for the OBT group. Mean change in HIV-1 RNA at week 24 was: -1.51, -1.86, and -1.68 for the 3 VCV dose groups, respectively; compared to -0.20 for the OBT group. Change in CD4 was +84, +142, and +142 for the 3 VCV dose groups at week 24, compared to 0 change for the OBT group. Change in Co-receptor use to X4/R5 or X4 only was 27% in the VCV 5 mg QD arm, 10% in the VCV 10mg QD arm, and 7% in the 15mg QD arm; compared to 4% in the OBT group.
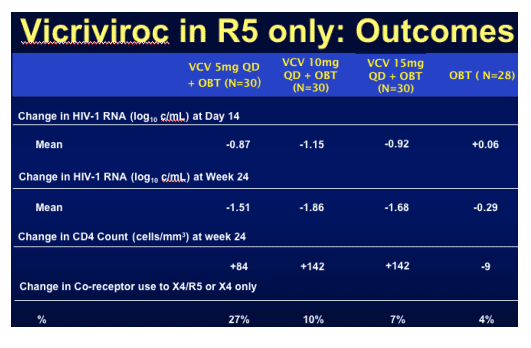
When the response was broken down by baseline tropism good suppression was seen even in the X4/R5 tropic populations.
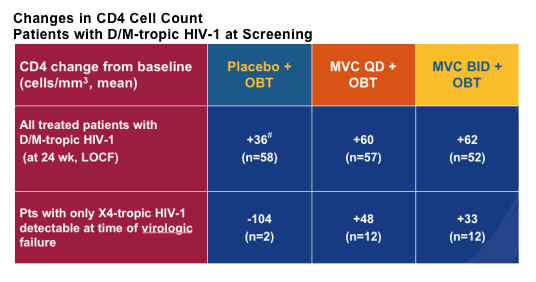
Howie Mayer from Pfizer showed the only new data on the CCR5 inhibitor maraviroc with the week 24 data from the A4001029 study [THLB0215]. This is an ongoing, double blind, placebo controlled study in subjects with triple-class failure, >5,000copies/mL and in those with non R5 virus, obviously a group of patients where you may expect the drug to have limited benefit. One hundred and ninety subjects were randomised to maraviroc 150mg QD or BD versus placebo, all with optimised background. Baseline data is shown below and most subjects were dual or mixed tropic. Around 50% received concurrent T20 and 60% in each group had between 2 and 4 active drugs in the background, baseline T4 was approximately 95 and viral load 5.0 log.
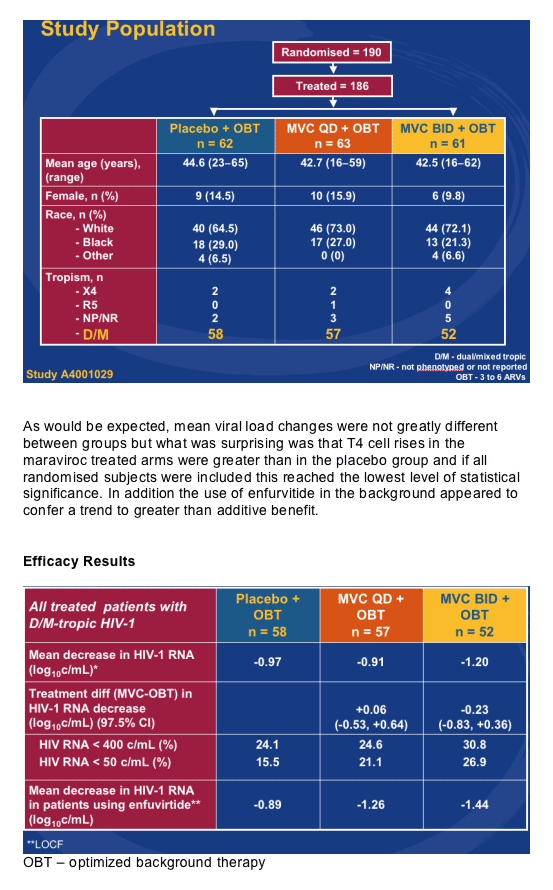
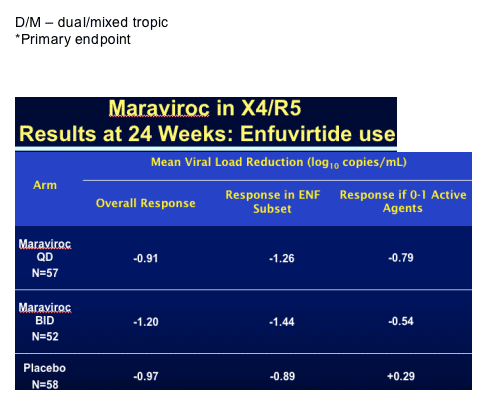
This raises the question of whether the drug may actually be useful in a wider range of patients than was first thought. In terms of toxicity nothing was seen to change the view that this is a well tolerated drug and specifically more grade 3 and 4 abnormalities in liver function were seen in the placebo group.
The new fusion inhibitor peptides, TRI-999 and TRI-1144 are being developed through a joint-venture between Trimeris and Roche an interaction which has already produced enfurvitide (T20). In a poster presentation the ability of these new compounds was examined to inhibit isolates resistant to T20 and the previous lead compound T-1249 [THPE0021]. Using an in vitro selection assay a series of mutations developed in both the HR1 domain of gp41 as well as mutations in the HR2 domain although these were less frequent. Whilst all the T20 resistance isolates showed less than 10-fold loss of susceptibility to T-1249, mutations arising through selection with T1249 were highly resistant to T20. When all these isolates were tested against either TRI-999 or TRI-1144 they retained sensitivity to these new agents. Both required higher numbers of mutations to show any resistance and virus breakthrough was only seen with concentrations of 30-fold less than T20 or T-1249, suggesting a much enhanced genetic barrier for resistance.
Barbara Del Medico then showed exciting data on the advances towards a once weekly sub-cutaneous formulation of the two lead compounds [THAA0303]. The aim was to develop formulations that result in minimum drug burst with maximum drug exposure. Using a rat model they first tried a peptide-organic salt complex which gave 13% bioavailability for TRI-1144 but a long half-life and an improved AUC when the release rate of compound was increased.
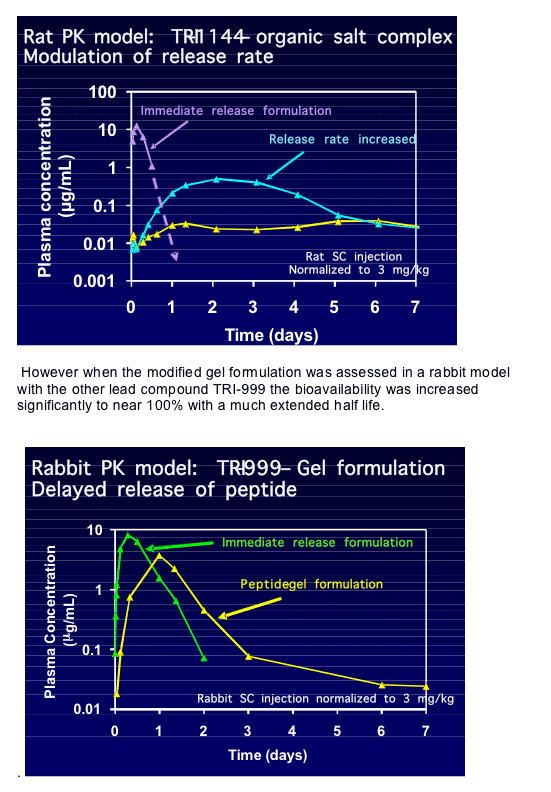
This is exciting as it shows that a once weekly injectable fusion inhibitor is possible and is a big leap towards the development of these compounds to the clinic which appears to overcome many of the current problems with T20. When asked if T20 was a candidate for the extended release formulation the presenter said that the PK profile did not lend itself to this and no work in this direction is planned. This makes the rapid development of these two new compounds all the more vital.
Pedro Cahn from the Fundacion Huesped in Buenos Aires showed data on a phase I study of the safety, tolerability and pharmacokinetics of fosalvudine tidoxil in HIV patients conducted in Argentina [THLB0216, THPE0025]. This agent is a pro-drug of the nucleoside RT inhibitor alovudine (FLT) and has a narrow therapeutic window. Four weeks at a dose of 7.5mg QD FLT has showed a median 1.88log drop in HIVRNA in a previous study in patients with multiple TAMs [Katlama et al, 2004]. Another early trial of FLT was stopped after a case of liver failure and at the end of the session the investigator in that study, Charles Flexner, commented that this was an idiosyncratic hepatic necrosis and that he advised investigators of this compound to be very careful! After the experience of lodenosine (2FDDA), where high rates of liver changes were seen, I would agree. Nevertheless the study (in 24 healthy volunteers) showed the pro-drug, thought to be safer since it has minimal bone marrow distribution and high protein binding, to have a half life of 5-7hours at doses from5, 10, 20 (molar equivalent of 7.5mf FLT) and 40mg single doses. Apart from headache no adverse events were reported.
Non-nucleoside reverse transcriptase inhibitor (NNRTI) data at the conference was exclusively from Tibotec on their two drugs TMC125 (etravirine) and TMC278 (rilpivirine). The only new information for the latter agent was a healthy volunteer study (n=16) to examine the interaction with ketoconazole, both an inhibitor as well as a substrate of the liver enzyme CYP3A4 [TUPE0087]. Ketoconazole increased the exposure to TMC278, itself a substrate of CYP3A4 by 49% (AUC24h), probably by inhibiting the metabolism of TMC278 within the liver. The study suggests that when TMC278 is combined with other inhibitors of cellular cytochromes that dose modifications may be necessary.
There were two pharmacokinetic studies with the other NNRTI, TMC125, which is nearer the clinic. There appears to be no significant interaction with this drug and methadone in healthy, HIV antibody negative volunteers, who are stabilised on methadone therapy [TUPE0084]. In a fourteen day study of sixteen individuals no clinically relevant changes in drug handling of either agent were seen and no significant symptoms of methadone withdrawal observed, which with the currently licensed NNRTI's can be a problem. A further bioavailability study of TMC125 was conducted, in 18 healthy HIV negative volunteers, assessing what happens when the agent is co-administered with the H2 antagonist ranitidine and the proton pump inhibitor omeprazole [TUPE0082]. Drug-drug interactions with the therapies used for ulcer pain and gastritis have been problematic for some other HIV drugs, notably unboosted atazanavir. In this case neither ranitidine nor omeprazole had any clinically important interaction, although TMC125 exposure was increased by 41% possibly due to the inhibition of CYP2C19.
Clinical data came from Cal Cohen reporting on the TMC125-C223 trial where 199 subjects with documented NNRTI resistance were randomised to TMC125 400mg or 800mg BID compared to standard of care [TUPE0061]. Baseline NNRTI resistance showed a mean of 2NNRTI mutations and a phenotypic mean fold change of only 1.7 for TMC125 compared to 41 for efavirenz and 61 for nevirapine. At week 48 the viral load reduction was -0.88 and -1.04 log for the two TMC125 arms compared to -0.14 log for the control arm, 78% of whom had withdrawn for virologic failure.
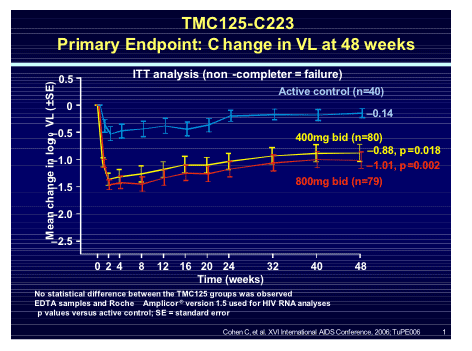
When the response was assessed against baseline resistance to NNRTI's the number of mutations predicted the response but even with 3 mutations the viral response to TMC125 remained greater than the active control.
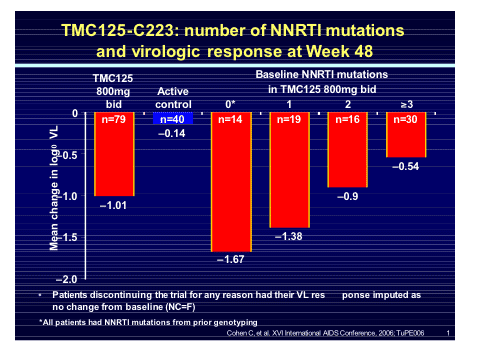
Following the PK study from Marta Boffito in London last year which defined the optimal dose to use as TMC114 600mg/100mg with TMC125 200mg BD this combination has entered into two large phase 3 studies (DUET 1 and 2) to assess the safety and efficacy.
Kakuda and co-workers from Tibotec reported a formal, two-way crossover trial to further assess PK interaction in 32 HIV-negative volunteers, randomized to two groups [TUPE0086]. All volunteers received 100mg TMC125 BID for 8 days. After a washout of 14 days, volunteers received TMC114/r 600/100mg bid for 16 days. From Day 9 to 16, group A received 100mg TMC125 BID; group B received 200mg BID.
Concomitant administration of TMC125 and TMC114/r had no clinically significant effect on TMC114 or ritonavir PK. TMC125 100mg BID exposure was decreased by 37% with similar decreases in Cmax and Cmin (32% and 49%, respectively). PK parameters for TMC125 observed after co-administration of TMC125 200mg BID with TMC114/r was slightly lower than historical control for healthy volunteers given the same dose and formulation (AUC12h 7,638�2254; Cmax 876�233).6. Overall, the safety profile for TMC125, TMC114/r, and their co-administration was similar. Apart from rash, which was seen more frequently during co-administration, use of TMC125 with TMC114/r in healthy volunteers was generally safe and well tolerated.
A number of groups around the World have early data on compassionate use groups receiving the two drugs simultaneously. One of the more clinically relevant studies for salvage therapy was a poster by Julio Montaner from Vancouver on the outcomes of combining TMC114/r with TMC125 [THPE0136]. In 4 heavily pre-treated subjects on this combination, 3 also received enfurvitide as a new drug, a drop of 2log was seen in HIV RNA and no adverse events were reported. This is similar to data we have from patients in London and this triple regimen offers a reasonable new HAART for those who cannot wait for novel classes such as integrase to be available.
MK-0518, the first integrase inhibitor developed Merck, is an agent that prevents HIV from incorporating its genetic information into that of the host cell and thereby prevents established infection of T cells. It has shown good efficacy in vitro and is synergistic with many available HIV drugs. Since it has a novel mode of action, within the nucleus of the cell, there should be no cross resistance and thus represents a vital new option for those with multi-drug resistance [THPE0020]. In addition favourable pharmacokinetics and metabolic properties of the agent suggest it will have a good safety and efficacy profile in HIV patients [THPE0027]. Michael Miller gave an update of the biochemical and antiviral activity of MK-0518 which charted the history of its development over the past 12 years, thanks to the tenacity of Daria Hazuda and her team at Merck [THAA0302]. The agent is highly specific for HIV integrase, is active against HIV-1 and HIV-2 and is either additive or synergistic with all tested antiretroviral agents although no data has yet been shown for tipranavir and darunavir. It is not expected, however, that there will be problem with using these agents with MK-0518. A question from the floor asked about the drug crossing the blood brain barrier and this has not yet been assessed.
Exciting new data on the drug was presented in a pivotal study in antiretroviral na�ve subjects [THLB0214] by Marty Markowitz (study design below).
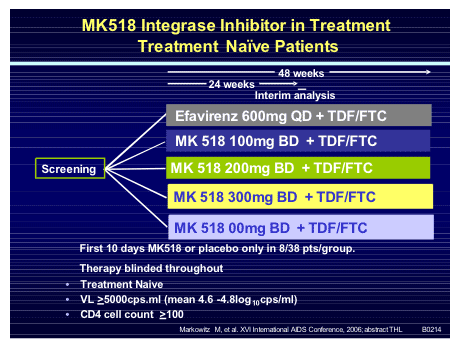
Considering the fact that efavirenz and Truvada have shown themselves to be very safe and highly effective in na�ve patients to pitch your drug against them in this patient group is somewhat of a risk and has been the Waterloo for several agents already (aplaviroc and vicriviroc). Thus it was comforting to see that MK-0518 performed well, especially since the 10 day mono-therapy lead in could also risk outcomes if resistance were to emerge. Enrolled subjects were mean age 36, 80% male, 69% non-white and 34% had AIDS.
In the graph immediately below, 89% & 88% in the 100mg arm had <400 & <50 c/ml, respectively, 100% in the 200mg arm had <400 & <50 c/ml, 100% & 94% had <400 & <50 c/ml in the 400mg arm, compared to to EFV arm in which 93% & 88% had <400 & <50 c/ml.
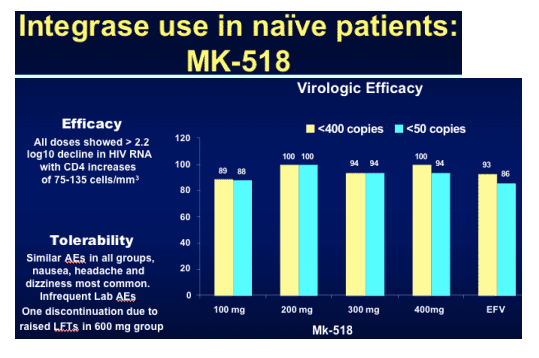
Not only did MK-0518 outperform efavirenz at all dose levels but also appeared to result in a greater proportion of patients undetectable at an earlier time point.
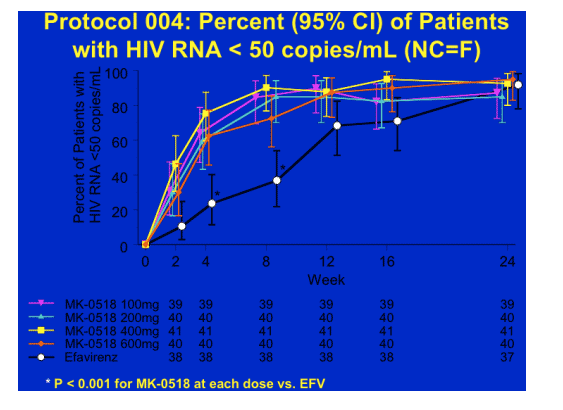
Finally the tolerability profile of MK-0518 was at lest as good as efavirenz, if not better and so the longer term follow-up of this study will be very interesting and potentially lead to a new option for patients na�ve to therapy.
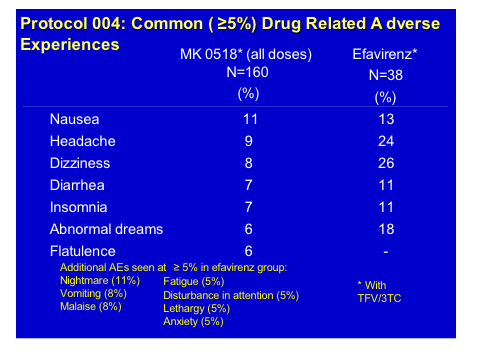
GS-9137 is the potent HIV-integrase inhibitor from Gilead which at a dose of 50/100mg ritonavir showed a >2.0log decrease in HIV RNA in a 10-day mono-therapy study recently published by Edwin De Jesus and co-workers [JAIDS 2006, 43(1):1-5]. At this conference there were two pharmacokinetic interaction studies, conducted by Brian Kearney and his group, to delineate if either zidovudine or Truvada (emtricitabine/tenofovir) had any meaningful interactions with this drug [TUPE0080, TUPE0088]. Since both zidovudine and GS-9137 are excreted renally through glucuronidation, although the latter is primarily cleared by the cytochromes in the liver, there was a potential for drug-drug interaction. In a standard cross-over design 28 subjects were given the highest planned dose of GS-9137/r of 200/100mg QD with 250mg of zidovudine. There appeared to be no clinically significant alteration of either drug level, co-administration was safe and well tolerated and the half-life of this dose of GS-9137, at 9.1h supports once daily dosing.
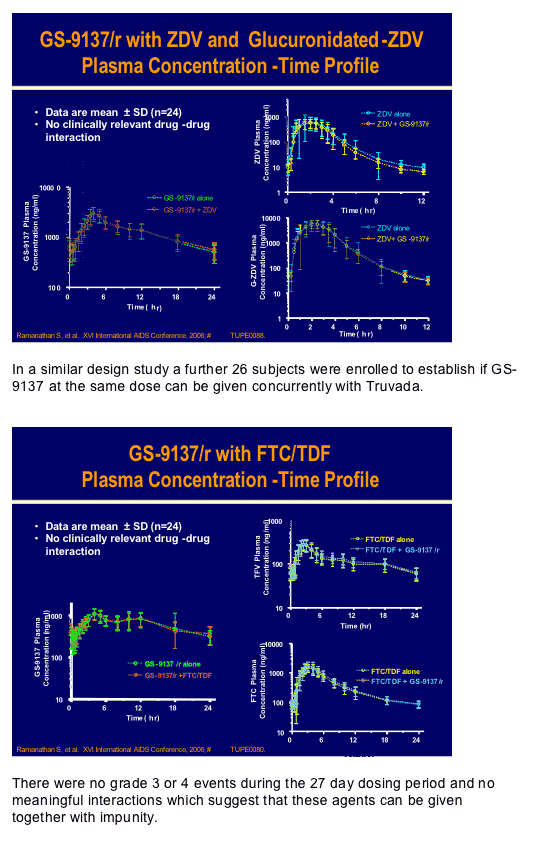
There were no grade 3 or 4 events during the 27 day dosing period and no meaningful interactions which suggest that these agents can be given together with impunity.
It was a mistake by the organisers of this meeting to have a session on new drug development at the same time as the late breaker presentations on the same topic, but for those of us who spent time in the former we were rewarded with news of an exciting new protease inhibitor, PL-100 [THAA0304]. Developed by Ambrilia Biopharm, in conjunction with Mark Wainberg, this agent is a potent, orally bioavailable PI with a simple synthesis process of only 5 stages. The precursor PPL-100 is converted into PL-100 which has a half-life of around 35hours and does not require the use of ritonavir boosting. In vitro studies selection studies produced viruses with reduced sensitivity to PL-100 (mutations selected were novel and included K45R, M46I, T80I and P81S) and at least 4 were required to produced a 10.8 fold reduction in sensitivity. There appears no cross-resistance to current licensed PI's and the T80I mutation results in hypersensitivity to saquinavir and nelfinavir. A phase 1 study was undertaken in healthy volunteers with doses of 300, 600, 1200 and 2400mg QD either fasted or with a light meal. Although food reduced Cmax it had no effect on AUC. No significant toxicity was seen (no grade 3 or 4 events) and the compound is now moving forward to clinical development.
The second PI moving towards the clinic Brecanavir for GlaxoSmithKline and data was presented by Charles Craig on susceptibility in vitro against a panel of 105 �worst case scenario� viruses from Monogram Biosciences a long with other viruses with single, double or triple mutations at positions (32, 33, 46, 47, 50, 54, 82, 84, and 90) [THPE0023]. Throughout this panel brecanavir retained activity at sub-nanomolar levels (median IC50:300pM) suggesting that increased numbers of resistance mutations will be required for the drug to fail. The presence of amino acid substitutions at protease residues 32, 47 and 50 were associated with the greatest reduction in sensitivity (6fold).
Interaction data for other PI's with brecanavir (BCV) from three multiple-dose drug interaction studies were shown doses of 300mg or 600mg with 100mg ritonavir boosting with a variety of PI's [TUAB0105]. In the first study with BCV 300mg and lopinavir/ritonavir no significant changes were seen in the levels of either agent and no unexpected adverse events were reported. In the second with BCV 300mg and atazanavir 300mg with 100mg ritonavir both agents showed increases in Cmax (by 38% for BCV and 48% for ATV) and AUC (41% for BCV and 21% for ATV). Some bilirubin changes were seen with atazanavir probably due to the increased drug levels.
For the final interaction data with tipranavir 500mg with ritonavir 200mg and BCV 600mg dosing was discontinued prematurely as 7 out of 12 healthy volunteer subjects developed increased ALT (Grade 1 (N=3), Grade 2 (N=2), Grade 3 (N=2) and this was thought to mean a high risk of poor drug-drug interaction. All these changes returned to normal after stopping the drugs. So in conclusion brecanavir seems best partnered form a PK perspective with Kaletra (lopinavir/r) and should not be dosed with tipranavir.
Of course the largest body of new data for PI's was for darunavir or TMC114 as the next drug about to be licensed in many countries. Fitting that most of the data was sliced from the pivotal POWER 1 and 2 studies augmented by POWER 3 all but one of the presentations were posters and several were combined analyses.
Starting with the clinical pharmacology, Sekar and co-workers from Tibotec presented an overview of all the studies which have led to the dose used of 600mg BID boosted with 100mg of ritonavir [TUPE0083]. In healthy volunteers, absolute oral bioavailability of TMC114 is increased by ritonavir boosting from 37% to 82% and increases systemic TMC114 exposure by approximately 14-fold. Giving a higher dose of 200mg of ritonavir does not increase TMC114 levels to any greater extent. TMC114 is 95% plasma protein-bound, mostly with alpha-1-acid glycoprotein (AAG) and its metabolism is almost exclusively by CYP3A4. A mass-balance study showed that TMC114 was excreted mainly in faeces (80%) and to a lesser extent in urine (12%) with a terminal half life of 15 hours. Although TMC114 exposure was approximately 30% lower under fasted than fed conditions, it was unaffected by meal type.
Tolerability data was shown for the three studies combined compared to those who received the comparator PI's (CPI) [TUPE0062]. There was a higher number of patient-years of exposure in the TMC114/r group (310) compared with the CPI group (75) due to the longer mean duration of treatment and the larger number of patients treated in the TMC114/r group. Few patients discontinued drug due to adverse events, 11% in the TMC114/r group and 81% in the CPI group compared to 3% and 67%, respectively, for virologic failure.
TMC114/r was generally well tolerated and no specific toxicity was associated with TMC114/r treatment. The incidence of diarrhoea was lower in the TMC114/r arm than in the CPI arm (16% versus 28%) as was headache (11% versus 20%). Around 29% of patients in each treatment group reported grade 3/4 adverse events and there was no difference between those seen with TMC114/r and the comparator group. Serious adverse events were seen in 15% of TMC114/r-treated patients and 14% of CPI-treated patients who reported at least one SAE, the most common being pneumonia and metabolic acidosis; no individual SAE occurred in >1% of patients. Eleven patients died in the TMC114/r arms. Causes of death were illicit drug overdose, acute and chronic pulmonary emboli, anal cancer, multiple organ failure due to sepsis, bacterial endocarditis, acute respiratory failure, meningoencephalitis, septic shock, chronic cryptosporidial diarrhoea, brain oedema and an unknown cause; all deaths were considered by the investigator to be unrelated to the study medication. One patient died in the CPI group 2 weeks after follow-up.
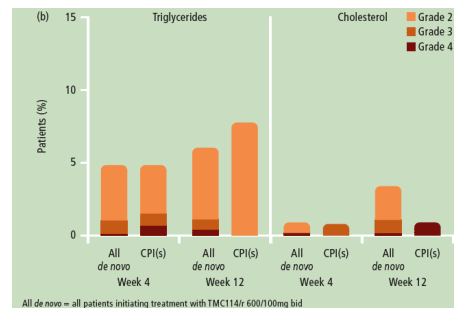
Coming on to laboratory abnormalities seen in the same population an integrated analysis was performed by Vangeneugden from Tibotec [TUPE0063]. The incidence of graded and non-graded laboratory abnormalities was generally low and similar between TMC114/r- and CPI-treated patients. Most were grade 1-2 and discontinuations due to these changes were uncommon (1% for both groups). The commonest grade 3 or 4 changes in the TMC114/r group were decreased white blood cell count (6% versus 7% in the CPI group), and increased triglycerides (9%), amylase (7%) and total cholesterol (5%) compared to 7%, 5% and 2%, respectively, in the CPI group)]. Changes in lipids over the first twelve weeks are shown below and show low rates of meaningful abnormalities over this short follow up, longer periods of therapy will be needed to clarify the comparative status of TMC114 in this area.
Finally a poster abstract, although I could not find it displayed, dealt with quality of life changes from POWER 1 and 2 and showed that using the validated Functional Assessment of HIV infection (FAHI) score showed clinically meaningful improvements were seen in the TMC114 arms (20-40% for various scores) compared to 15-25% in the CPI group [TUPE0069].
Two posters dealt with data for POWER three alone which was the follow-up study in which all subjects received TMC114/r 600/100mg from baseline. POWER 3 was non-randomized, open-label and enrolled 327 patients, 75% Caucasian, 87% male with a mean entry viral load of 4.62log and T4 count of 115 cells/mm3. To assess if there was any relationship between drug exposure and efficacy or safety week 4 trough and peak PK samples were taken and in addition the baseline resistance measurement was used to calculate the inhibitory quotient (IQ) of the drug, i.e. how much drug is needed to deal with the amount of resistance that exists to the drug [TUPE0078]. It appears from these data that TMC114 IQ was a strong predictor of virologic outcome, which is not surprising, and that most of this is driven by the baseline resistance fold change to the drug and not TMC114 exposure levels. With regard to safety however there seemed to be no association of TMC114 drug levels with any adverse events and this is comforting that signature toxicity does not seem to be a hallmark of this compound. The second POWER 3 analysis presented by Jean-Michel Molina showed results to be broadly similar to the first two studies with 65% having a >1log drop in HIV RNA by week 24, 40% reaching <50 copies/mL and a mean VL reduction of -1.65log [TUPE0060]. The most common adverse events were diarrhoea (14%), nasopharyngitis (11%) and nausea (10%). Grade 3 or 4 triglyceride, cholesterol, and liver function enzyme elevations occurred in 6%, 4%, 2% and 2% of patients, respectively, similar rates to these seen in previous TMC114/r studies.
Finally Sharon Walsmley from Toronto gave an oral on the week 48 combined analysis of POWER 1 and 2 where 241 subjects treated with TMC114 were available to be compared with data from 244 who received comparator PI regimens [TUAB0104]. Ninety one TMC114 subjects were <50copies/mL compared to 22 given CPI (p<0.001) and the breakdown by baseline subgroups is shown below.
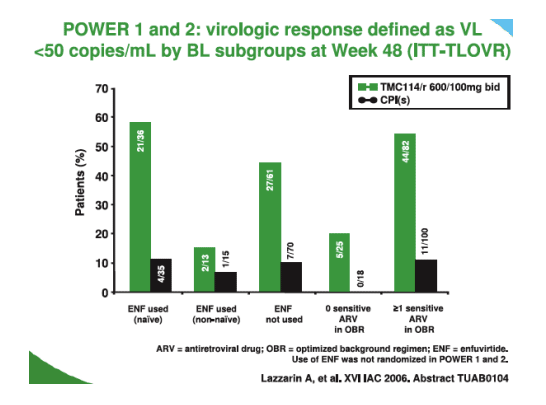
Clearly the use of enfurvitide within the back group is important for those patients still sensitive to the agent. T4 cell responses were good in the TMC114 groups 92/102 compared to the CPI 17/19; p<0.001 and P<0.05 respectively for Power 1 and 2). Adverse events were similar to those seen earlier with around 20% having some diarrhoea and 15% headache; the majority of adverse events were grades 1 and 2. A word of caution is that it is always difficult to assess side effects in this group of late stage patients since immune reconstitution can produce many constitutional symptoms also.
All in all a hopeful set of presentations putting into perspective the widening gap which will occur between those who have access to good drugs and those that do not. However, despite the huge advances that have occurred to give us new therapies to use it remains vital that doctors and health care professionals know how to use these therapies wisely to slow avoid the emergence of resistance which has been the hallmark of poor clinical practice in the past.
Thank you for those investigators who shared slides from their presentations.
Mike Youle
|
| |
|
|
|
|
|