 |
|
|
| |
Summary from CROI 2015 for Hepatitis C
HIV/HCV coinfected patients are no longer a difficult to treat special patient population.
What can we learn from new clinical phase III studies and feedback from real-life cohorts receiving modern DAA-based therapy?
|
| |
| |
Jurgen K. Rockstroh M.D., Professor of Medicine
University of Bonn, Germany
Correspondence:
Prof. Dr. J.K. Rockstroh
Department of Medicine I
University of Bonn
Sigmund-Freud-Str. 25
53105 Bonn
Germany
Introduction
At this year CROI, viral hepatitis once again, represents one of the major topics with new data from phase-3 clinical studies in coinfection, as well as feedback from implementation of DAA-based therapy in real-life HIV/HCV cohorts being presented. Also very interesting, new insights into practical aspects such as drug-drug interactions between HIV and HCV drugs as well as value of viral load monitoring under HCV therapy have been discussed at this meeting. Also some updated new information from treatment trials in HCV mono-infection was shown. Finally, in the HCV symposium on Thursday issues around HCV drug pricing and treatment access as well as HCV treatment as prevention were discussed. After all, HCV cure rates of >95% are fantastic to have but still need to be brought to the patients in need. This includes improving testing strategies for HCV as well as decreasing drug costs to make these therapies available for all.
New data from phase III trials in HIV/HCV coinfection
ION-4
The ION-4 study is a phase 3, multicenter, open-label study which was conducted in US, Canada and New Zealand and included genotype 1 or 4 patients who were either HCV treatment naïve or treatment-experienced. 20% of patients were allowed to have compensated liver cirrhosis upon inclusion (1). Cutoff inclusion criteria for platelets were ≥50,000/mm3, hemoglobin ≥10mg/dl, and CrCl ≥ 60mLmin. HIV Patients had to be well controlled for HIV with an HIV-RNA<50 copies/ml and a CD4 cell count above 100 cells/mm3. Patients could either be on rilpivirine, efavirenz or raltegravir in combination with TDF/FTC. No boosted PIs were allowed into the study as PK interactions studies had not been completed upon initiation of the trial yet. The study design is depicted in figure 1.
Figure 1: Study design of ION-4
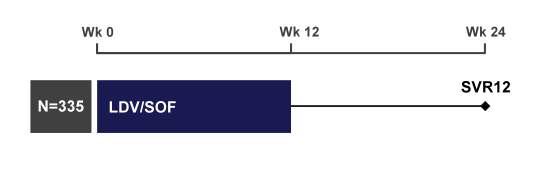
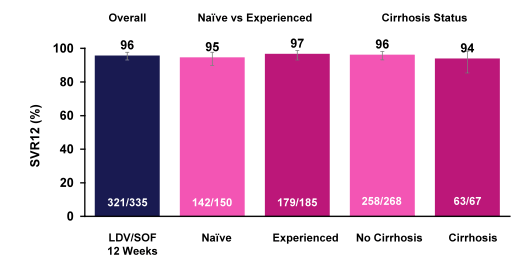
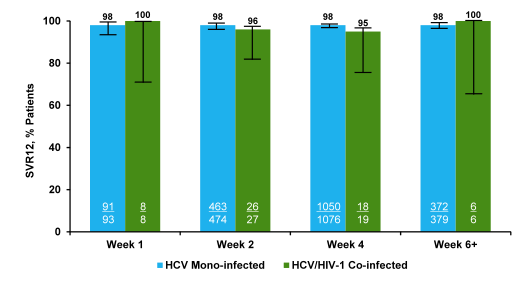
Safety evaluations included adverse event (AE) and standard laboratory parameter monitoring in addition to enhanced renal toxicity monitoring, CD4 count and HIV-1 RNA levels. The primary efficacy endpoint was SVR12. Overall, an impressive 335 patients with GT1a (75%), GT1b (23%) and GT4 (2%) were enrolled; 82% were male, 61% were white, mean age was 52 (range 26-72), mean baseline HCV RNA was 6.7 log10 IU/mL (range 4.1-7.8), median baseline CD4 count was 662 cells/uL (Q1, Q3=469, 823), 20% had cirrhosis, 24% were IL28B CC genotype and 55% had not responded to prior HCV treatment. Patients were taking efavirenz (48%) or raltegravir (44%) or rilpivirine (9%). The figure 2 shows overall SVR12 rate as well as SVR12 results according to HCV treatment history and presence of cirrhosis at baseline. Reassuringly, the overall SVR12 rate was 96% (321/335); no difference was seen in patients with cirrhosis or treatment failure underlying the robustness of this regimen. Only 2 patients had on-treatment virologic failure most likely due to non-compliance and 10 had virologic relapse after discontinuing treatment.
Figure 2: SVR12 rates in ION-4
In multivariate analysis black race emerged as the only predictive factor being significantly associated with a lower SVR rate. No clear explanation at this point can be made for this observation. In the question time following the presentation at CROI differences in drug levels or adherence were ruled out as possible explanations. It was also pointed out that no racial impact was found in any other of the ION studies. Therefore further pharmacogenomics investigations are being planned to further explore this finding.
No patient had confirmed HIV virologic rebound and no patient discontinued therapy because of adverse events. As ledipasvir may increase tenofovir exposure (potentially because of persistent inhibition of efflux drug transporters) careful renal monitoring was carried out during the trial. Indeed 4 patients (1%) within the trial had changes in creatinine ≥ 0.4 mg/dL of whom 2 completed treatment with no ART change, 1 had a dose reduction of TDF, and 1 discontinued TDF. Even more increases in tenofovir levels can be expected when ledipasvir/sofosbuvir is co-administered with boosted HIV protease inhibitors as they themselves can further increase TDF exposure. This is important to keep in mind when selecting HIV therapy when planning ledipasvir/sofosbuvir therapy.
In conclusion, ledipasvir/sofosbuvir for 12 weeks achieves impressive SVR rates which are very similar to treatment outcome reports from studies with this DAA combination in HCV mono-infection. Patients on tenofovir need careful renal monitoring as ledipasvir may increase tenofovir exposure. Clearly this interferon and ribavirin free regimen promises great tolerability with not one single discontinuation because of an adverse event in this relatively large trial.
CROI: Ledipasvir/sofosbuvir for 12 Weeks in Patients Coinfected With HCV and hiv-1: ION-4 - (02/27/15)
Ally-2
ALLY-2 is the phase III study of daclatasvir (DCV) + sofosbuvir (SOF) in patients with HIV/HCV coinfection (2). This randomized, open-label study enrolled HCV treatment-naive (N=151) or experienced (N=52) adults coinfected with HIV and HCV (any genotype). Naive patients were randomly assigned (2:1), with stratification by cirrhosis status and HCV GT, to receive 12 or 8 weeks of once-daily SOF 400mg + DCV 60mg (dose-adjusted for concomitant antiretrovirals: 30mg with ritonavir-boosted PIs, 90mg with NNRTIs except rilpivirine). Of note this is the first study to look at shorter treatment durations with DCV + SOF in HIV coinfected individuals. Experienced patients received this same regimen for 12 weeks. The primary endpoint was HCV RNA < LLOQ (25 IU/mL) at post-treatment Week 12 (SVR12) in naive GT1 patients treated for 12 weeks. The study design is shown below in figure 3.
Figure 3: Study design of ALLY-2
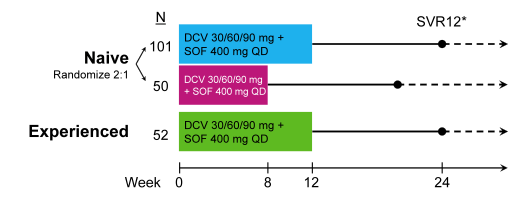
Treatment arms were well balanced with a median age of 52 y, 87% male, and 62% white/34% black. 83%, 9%, 6%, and 2% of patients, respectively, had GT1, 2, 3, and 4 infection. The median baseline HCV RNA was 6.7 log10 IU/mL and overall 14% of patients had cirrhosis. The median baseline CD4 count was 565 cells/μL and 94% had HIV RNA <50 cp/mL. 99, 50, and 50 patients, respectively, received PI-based, NNRTI-based, or other (primarily INI-based) cART; 4 patients did not receive combination antiretroviral therapy (cART). Of note, the different antiretroviral therapies were not balanced between the different treatment arms. In the naïve 8 week arm the highest number patients receiving darunavir/r (42% vs 19% and 21% in the 12week naïve and experienced arm, respectively) were included. Overall, 199/203 (98%) patients completed therapy; 1 patient discontinued early for incarceration. The main efficacy results are depicted in figure 4.
Figure 4: SVR12 rates for GT1 and the overall patient population in Ally-2
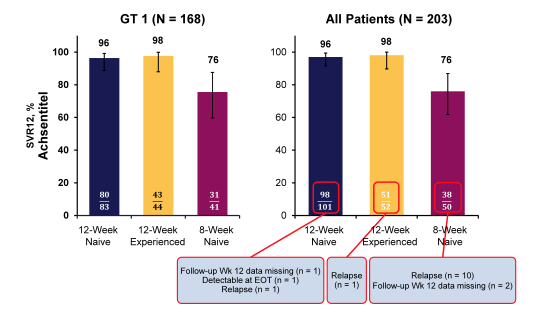
Overall, SVR12 was achieved by 97% and 98% of naive and experienced patients, respectively, after 12 weeks of therapy, but only 76% of naive patients after 8 weeks. Among patients treated for 12 weeks, SVR12 rates were similar regardless of prior treatment experience, HCV GT or GT1 subtype, cirrhosis status, concurrent cART regimen, or race. There were no virologic breakthroughs; 2/153 patients (1%) in the 12-week group and 10/50 (20%) in the 8-week group had post-treatment relapse. The observed higher relapse rate in the 8 week arm raises the question whether 8 weeks are to short and whether predictive factors can be identified allowing to foresee who would be sufficiently treated with the shorter treatment duration. One difficulty in analysing the data is that over 40% of the patients from the 8week arm were on concomitant darunavir/ and therefore only dosed with 30mg of daclatasvir. This was originally done on the basis of interaction studies with atazanavir/r where significantly increased daclatasvir levels were observed warranting a daclatasvir dose reduction. More recently however, data was presented in December 2014 from the HIV DART meeting showing that daclatasvir had fewer interactions with boosted darunavir or lopinavir so that nowadays no dose reduction is recommended (3, see link below to data). This implies though that lower daclatasvir exposure in the darunavir treated patients may also contribute to higher relapse rates. At the time of this presentation however, no clarifying drug level analyses were available. The fact that ART choice overall in the 12week arms had no impact on SVR rates though makes this very unlikely.
There were no treatment-related adverse events leading to treatment discontinuation. Treatment-emergent grade 3/4 lab abnormalities included increases in INR (2 patients), AST (1), total bilirubin (8, all received ATV/r), and lipase (7, all transient without pancreatitis).
In conclusion, the interferon-free and ribavirin-free DAA combination of daclatasvir and sofosbuvir for 12 weeks achieved very high cure rates in HIV/HCV coinfected subjects patients coinfected with HIV and predominantly HCV GT1 infection. Tolerability of this regimen was very good with no treatment related discontinuations.
Unfortunately, both ION-4 (included GT1 and 4) and Ally-2 (included GT 1,2,3 and 4) included too little on other genotypes to allow final conclusions on the efficacy in these specific genotypes. Nevertheless, it would be helpful to see subanalysis looking at response by genotype.
CROI: Daclatasvir in Combination With Sofosbuvir for HIV/HCV Coinfection: ALLY-2 Study - (02/27/15)
HCV/HIV DART: Daclatasvir: Overview of Drug-Drug Interactions With Antiretroviral Agents and Other Common Concomitant Drugs - (12/16/14)
Other new trial information
For the first time results from an ANRS open label, single arm, phase 2 study (ANRS HC30 QUADRIH) on daclatasvir/asunaprevir/PEG-IFN/RBV in HIV/HCV genotype (GT) 1 or 4 co-infected patients with previous null response to prior pegylated interferon/ribavirin therapy was presented in the oral hepatitis session (4) Now obviously the PEG-IFN component makes it unlikely that this regimen will play a role in the future but it does present the first data in coinfection with the BMS daclatasvir/asunaprevir combination. All patients received a 4-week lead-in phase with PEG-IFN/RBV, followed by 24 weeks of asunaprevir (100mg bid), daclatasvir (60 mg qd), and PEG-IFN/RBV. The primary endpoint was sustained virological response 12 weeks after the end of treatment (SVR12) using ITT analysis. Seventy-five patients (59 men/16 women) were included, median age was 50 (IQR: 48 - 53) years, and 27 (36%) were cirrhotic. All were on a raltegravir-based regimen, 92% with baseline plasma HIV RNA level <50cp/ml and median CD4 count 748 (481 - 930)/mm3. The SVR12 rate was 96.0% (72/75; 95%CI: 91.6%-100%) for the entire study population. SVR12 was 92.6% (25/27; 82.7%-100%) in cirrhotic patients, 94.6% (35/37; 87.3%-100%) in GT1and 97.4% (37/38; 92.3%-100%) in GT 4. Six patients (8%) prematurely stopped HCV therapy, 2 due to virological breakthrough, and 4 to adverse events (one lung cancer, 3 infections). Overall, 35 serious adverse events occurred in 21 (28%) patients (9 cirrhotic and 12 non cirrhotic, p=0. 44), including hematological (15%, mainly anemia and neutropenia), gastrointestinal (5%), psychiatric events (5%, mainly insomnia), and infections (5%). In conclusion, the combination of regimen study was very efficacious but the high rate of serious adverse events in this cirrhosis enriched patient population underlines the need for IFN-free HCV treatment options in patients with advanced liver disease.
CROI: SVR12 in over 90% of HCV/HIV null responders stating daclatasvir/asunaprevir plus PR - (03/02/15)
Feedback from real-life patient cohorts
An entire themed poster discussion session on Thursday dealt with treatment outcome results of first mostly interferon free DAA-based HCV therapies in HIV/HCV coinfected subjects from various clinics and cohorts around the world.
In a German study real-life data on sofosbuvir-based treatments was presented (5, poster). In this multicenter cohort, all patients who were started on the following treatment regimens were documented: sofosbuvir (SOF) plus ribavirin, SOF/daclatasvir, SOF/simeprevir, and SOF/PegIFN/RBV. In total 518 patients have been enrolled so far. Of those, 393 were HCV-monoinfected and 125 HCV/HIV-coinfected. Liver cirrhosis was present in about a third of the patients. Slightly more than half of the patients were pre-treated for HCV. Most HCV/HIV patients had well controlled HIV infection and a high CD4+ cell count. Only 1/125 HCV/HIV-coinfected patients was not on antiretroviral therapy. For the current analysis presented at CROI due to the limited number of HCV/HIV-coinfected patients with HCV genotype 2 and 3 only patients with genotype 1 and 4 were analysed. For patients with HCV genotype 1 or 4 there was no difference in virologic response between SOF/PegIFN/RBV, SOF/SMV or SOF/DCV. In addition, response rates for HIV/HCV-coinfected and HCV-monoinfected patients regardless of the SOF-containing regimen were similar. This again supports the current EASL and AASLD guidelines which no longer separate between HIV/HCV coinfected and HCV monoinfected patients in the DAA era. Also on average real-life SVR rates were either similar or only slightly lower than what has been reported from controlled clinical trials. Lower SVR12 rates were particularly seen in patients with cirrhosis. Premature discontinuation or lost to follow up were observed in 5% of patients counted as treatment failure, whereas relapse occurred in 7%. Figure 5 summarizes the main findings of the study.
Figure 5:
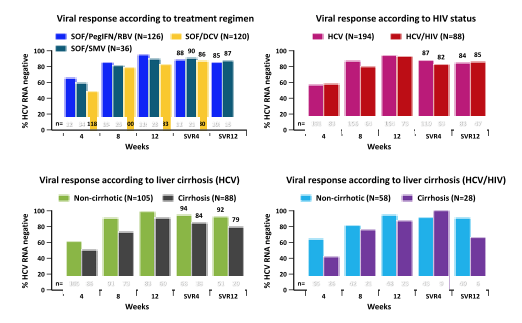
In conclusion, in this ongoing cohort with a substantial proportion of patients with advanced fibrosis, liver cirrhosis but not HCV/HIV-coinfection seems to be associated with a lower SVR rate. Premature discontinuation and lost to follow up, compared to relapse, almost equally contributed to patients considered as treatment failure.
Additional studies assessing sustained virologic responses and tolerability of sofosbuvir + simeprevir in HIV/HCV-coinfected patients alone or in comparison to those with HCV mono infection were presented from different sites from the US.
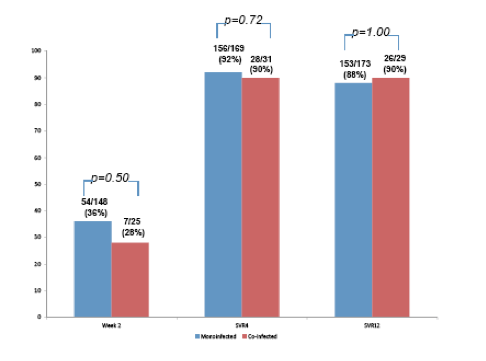
In a study from Mount Sinai Hospital starting in December 2013 all patients with chronic HCV who received either simeprevir and/or sofosbuvir were analysed (6, poster). Overall, records of 594 HCV-positive patients were reviewed. Data was collected on 78 HIV-positive patients with chronic genotype 1, 2 or 3 HCV infections who initiated: SMV/SOF with or without RBV or SOF/RBV. In this study, advanced fibrosis/cirrhosis was defined by biopsy and/or a FibroScan score ≥ 13.5 kPa and/or a FIB-4 score ≥ 3.25. Week 2, SVR4 and SVR12 rates among the co-infected patients were compared to those HCV mono-infected patients placed on SMV- and/or SOF-containing regimens during the same period. 56% of the coinfected patients were HCV treatment experienced and 52% had advanced fibrosis. No difference in week 2, SVR4 and SVR12 were observed between co- and monoinfected patients on SMV/SOF ± RBV (see figure 6). These observations again strengthen guideline recommendations to no longer separate co- and mono-infected patients from each other.
Figure 6: Week 2, SVR4 & SVR12 Results for HCV versus HIV/HCV Patients on SMV/SOF ± RBV
Additional work was presented from Mount Sinai evaluating patients on sofosbuvir (SOF) and/or simeprevir (SMV) experiencing hepatic decompensation and serious adverse events (SAEs) in a real-world setting (7, poster). For data analysis, the study group was comprised of patients who had not undergone liver transplantation (LT) (Cohort 1, which included 499 patients), and patients who had already undergone LT (Cohort 2, which included 45 patients). In cohort one 4.5% of patients experienced liver decompensation or an SAE during treatment or within one month after end of treatment (EOT). The untoward events were likely to be treatment related because the cases had not had similar events for at least the 12 month period prior to starting treatment. Three cases died unexpectedly with no underlying conditions considered to be life-threatening. One patient was Child Pugh Class B who was prescribed SMV/SOF. The overall mortality was 0.6%. Of the 13 surviving patients, four relapsed after EOT, one was viral load undetectable at EOT, two were viral load detectable at the time of treatment discontinuation, and six achieved SVR 12. Liver decompensation or an SAE led to treatment discontinuation in 1.4% (7/499). Risk factors for decompensation/SAE included low baseline albumin and high total bilirubin. Interestingly, fibrosis stage was not a risk factor which may have been related to the overall high percentage with more advanced liver disease or cirrhosis. The 4.5% incidence of decompensation/SAE suggests that a subgroup may exist who could benefit from more intensive monitoring or some other type of intervention. In cohort two 28% of patients experienced liver decompensation or an SAE during treatment or within one month of ending treatment. Liver decompensation or an SAE led to treatment discontinuation in 4.4% (2/45) of patients. Low baseline hemoglobin was identified as a risk factor for hepatic decompensation/SAE. In summary, low hepatic reserve may have increased decompensation risk in the non-LT patients. Based on past and current data, SMV should not be used in Child Pugh Class B and C patients. The underlying mechanisms leading to life-threatening adverse events or decompensation from SOF- and/or SMV-containing regimens need to be investigated further.
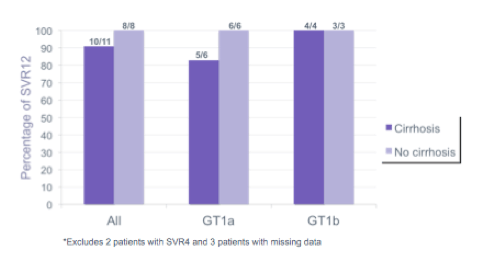
A study from the Northwestern University Viral Hepatitis registry (NU-VHR) assessed the safety, efficacy and tolerability of selected oral DAA combinations in 44 HIV/HCV co-infected enrolled in the NU VHR, a prospective observational cohort of ambulatory HIV-infected patients co-infected with chronic hepatits B or C (8, poster). 54.5% of the patients treated were cirrhotic, 63.6% GT1a, 27% 1b, 4 patients GT 2 or 3 (1), approximately half of the cohort were Caucasian. Of the 33 patients started on SOF/SMV, 29 patients have completed therapy so far. 27 patients were observed to have an end of treatment response, 2 patients were missing data at this time point. 18/19 patients who were followed to 12 weeks after completion of therapy achieved SVR. 3 patients had no data available for this time point. 1 relapse was reported. no virologic breakthroughs occurred. In conclusion, overall SVR rate within this on-treatment analysis was 91% in cirrhotics and 100% in non-cirrhotics. Clearly, these findings underline that study results from phase 3 trials can be reproduced in sicker real-life patient cohorts. [From Jules: authors say in poster: "Closer monitoring of a dually infected population and a multi-disciplinary approach to treatment may account for our higher success rates."]
Colleagues from Pennsylvania performed a cohort study among HCV-infected patients treated with sofosbuvir/simeprevir at 4 community-based and academic centers (9). The main outcome was end-of-treatment (EOT) HCV virologic response. HCV RNA, liver aminotransferases, and sofosbuvir/simeprevir discontinuations were evaluated over 12 weeks of treatment and 12 weeks of follow-up. Results were stratified by HIV status and by the presence of advanced hepatic fibrosis/cirrhosis. Overall, 81 patients (37 coinfected; 44 monoinfected) were treated with sofosbuvir/simeprevir between 12/2013 and 9/2014. Fifty-nine percent were African American, 61% were male, 73% had METAVIR stage 3/4 fibrosis, and 46% had prior HCV therapy (49% null or partial responders; 16% relapsers; 35% stopped due to toxicity). The most common HIV regimens in coinfected persons included raltegravir, dolutegravir, or rilpivirine with either tenofovir/emtricitabine or abacavir/lamivudine. Among HIV/HCV patients, 54% and 87% achieved an HCV RNA that was not quantifiable at 2 and 4 weeks of therapy, respectively, compared to 55% and 81% for HCV-monoinfected patients (p>0.5). Those with METAVIR stage 3/4 were equally likely to achieve HCV suppression by 4 weeks compared to those with less fibrosis regardless of HIV status (61% vs. 67% in coinfected patients, p=0.68; 56% vs. 75% in monoinfected patients, p=0.33). EOT and SVR12 results were also similar between the 2 groups and are shown in figure 7.
Figure 7: Virological response to sofosbuvir/simeprevir in HIV/HCV coinfected subjects versus HCV mono-infected subjects.
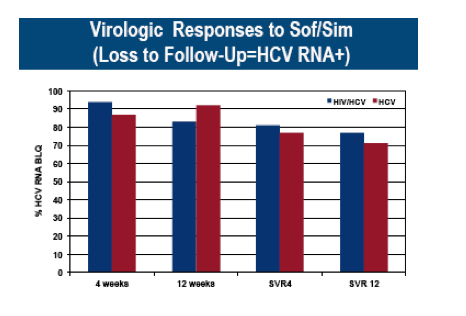
In conclusion, an all-oral sofosbuvir/simeprevir regimen was effective for patients with chronic HCV genotype 1, regardless of HIV status, previous treatment response, and stage of fibrosis. Adverse events were rare, even in patients with advanced fibrosis/cirrhosis.
CROI: Effectiveness of Sofosbuvir/Simeprevir for HIV/HCV Patients in Clinical Practice - (03/09/15)
Interestingly, there was also data presented from a small group of prior HCV PI treatment failures who were retreated with sofosbuvir and simeprevir ± ribavirin (10). These genotype 1 patients were treated because of advanced stages of liver disease and immediate clinical need. Patients were characterized by high rates of cirrhosis, predominance of genotype 1a, and high HCV RNAs as you would expect from a cohort of patients failing PIs. Patients received a PI a median of 29 months prior; however some had received PIs several years prior. Telaprevir was used most commonly, but boceprevir and other PIs were also used. Most of the patients were non-responders to HCV-PI defined for the purpose of this study as failing to suppress or breaking through during the period of PI treatment. All patients received testing for HCV resistance prior to SOF+SIM based treatment. Overall, no mutations associated with significant PI resistance were found at baseline but 3/9 genotype 1a viruses had the Q80K polymorphism. Two thirds received RBV with SOF+SIM. By week 4 on treatment all patients were below limit of quantification (BLQ) while 60% were below limit of detection (BLD). At EOT all patients were undetectable and all but 1 remained so 12 weeks after stopping therapy (SVR12). In summary, SOF+SMV+/-RBV treatment may be appropriate for carefully selected PI-experienced G1 pts including those with cirrhosis and as predicted by the genotypic testing, SMV appeared to have activity in the regimen since much lower SVR rates would have been expected without it. Clearly the long time period between first PI-based virological failure and potential resistance development and new retreatment may have played a crucial role in obtaining the high SVR rates observed in this pilot study. [from Jules: in the abstract & in speaking with author PI treatment occurred 5-88 months prior so some patients had genotype testing soon after failing & none of these patients treated with Sim/sof in this study had SMV genotypic mutations detected.]
CROI: Sofosbuvir, Simeprevir, +/- Ribavirin in HCV G1 Protease Inhibitor-Experienced Patients [14/15 SVR 93%] - (03/09/15)
Clinically important information derived from a study which assessed the impact of drug-drug interactions (DDIs) between Simeprevir (SMV) and antiretrovirals (ART) in limiting HCV treatment among HIV/HCV co-infected individuals in clinical practice settings (11, poster). The need to switch antiretroviral therapy (ART) prior to initiation of SMV and the feasibility of switching ART to allow SMV use were studied through a retrospective chart review at the University of Pittsburgh Medical Center's HIV/AIDS Program from June-August 2014. All patients with HIV and chronic HCV with a visit in the past 18 months were included. After collection of baseline characteristics, significant interactions between SMV and ART were identified based on available literature. If DDIs limited use of SMV, previous HIV genotype reports were reviewed to determine the feasibility of a safe and effective ART switch. Of 133 patients, 71% were male, 54% African American, 23% met criteria for advanced liver disease, 86% had HCV genotype 1, and 94% were currently on ART. The distribution of regimens was: ritonavir-boosted PI (PI/r) (38%); efavirenz (34%); raltegravir (11%); rilpivirine (6%); elvitegravir/cobicistat (1%); and other regimens including dolutegravir (4%). An ART switch to allow use of SMV was required in 103 (77%), most frequently for patients on efavirenz or a PI/r. For 47 (46%), a straightforward substitution could be made. For the remaining patients, a switch following HIV expert opinion was viable in 40 (39%), but no switch was possible in 16 (15%) due to archived HIV drug resistance mutations. Notably, for more than 30% of patients on a PI, an ART switch was not feasible. In summary, the majority of HIV/HCV co-infected patients will require ART switch prior to use of SMV. Additionally, for nearly a third of patients on a PI, an ART switch may not be feasible. These findings are significant to real world clinical practice settings and highlight the complexity of using interferon-free DAAs in the coinfection setting. Although these seems most pronounced in HCV PIs this still also holds true for non PI based DAA combinations.
Finally, first real-life patient cohort data was presented from a small observational cohort of HIV/HCV coinfected patients from the University of Nice in France who received sofosbuvir (SOF) and daclatasvir (DCV) combination therapy (12, poster). In this study the safety and efficacy of SOF plus DCV and plasmatic concentrations of antiviral drugs in HCV/HIV co-infected patients was evaluated. HCV patients with extensive liver fibrosis (METAVIR F3 and F4) and stable HIV disease received SOF 400 mg QD and DCV (30, 60 or 90 mg QD) for 24 weeks. Residual plasma concentrations of DCV, sofosbuvir's metabolite (GS331007) and ongoing ART were determined on patient's blood samples 15 days after starting HCV treatment. The primary efficacy endpoint was sustained virologic response 12 weeks after treatment discontinuation (SVR12). Overall, 26 patients were included into the analyses. 77% were male, median age was 52 (IQR 48-62), 96% had an undetectable HIV viral load, 80% were genotype 1, 81% had F4 fibrosis and 84.6% had previously failed an IFN/RBV combination therapy. HCV viral load under treatment was undetectable in 5/26 (19%) at Week 2 (W2), 12/25 (48%) at W4, and 20/20 (100% ) at W24. In a per-protocol analysis 93% of patients achieved SVR4 and 86% SVR12, respectively. The PK-substudy showed no large inter-individual variability of DCV Ctrough, strengthening the data suggesting no dose adjustment requirement in patients with any degree of renal impairment or mild-to-severe hepatic impairment. In the study daclatasvir dose was adjusted according to HIV therapy (30mg for boosted PI/r and 90mg for efavirenz). In the study little impact of HIV drugs on DCV Ctrough was observed. On the other hand, significant outliers for GS-331007 Ctrough were noted. Previous studies showed that while SOF AUC0-24 is highly impacted in patients with moderate and severe hepatic impairment compared to subjects with normal hepatic function, GS-331007 AUC0-24 is poorly affected. In contrast, mild and moderate renal impairment could affect the GS-331007 AUC. In the present study, renal function impairment did not appear to contribute to the significant outliers observed with GS-331007. SOF plus DCV combined therapy did not decrease raltegravir and darunavir Ctrough below target values. In summary, high SVR rates were obtained with the sofosbuvir/daclatasvir combination. No alarming PK values were obtained despite lower daclatasvir dosages with boosted HIV PIs.
PK-interaction studies: anything new?
At this year CROI data was presented on the interaction between the ledipasvir/sofosbuvir fix dose combination and commonly used HIV antiretrovirals (13). Most importantly the interaction between ledipasvir and tenofovir in particular in combination with a boosted PI was examined. Indeed, increased tenofovir levels have been described under ledipasvir therapy. As tenofovir levels are also enhanced under boosted PI treatment the question was therefore how high the TDF levels may become and whether this would potentially increase the risk for renal toxicities. This was a multiple-dose, randomized, cross-over study in healthy volunteers. In Part A (simultaneous dosing), subjects received LDV/SOF, ARVs (Cohort 1: ATV/r (300 mg/100 mg)+TVD (200 mg/300 mg); Cohort 2: DRV/r (800 mg/100 mg)+TVD), and LDV/SOF+ARVs each for 10 days. In Part B (Cohort 3 and Cohort 4), an evaluation of staggered (12 hour) dosing of LDV/SOF and ARVs was conducted. LDV, SOF, GS-331007 (predominant circulating metabolite of SOF), and ARV plasma concentrations were analyzed and PK parameters were calculated. 90% CIs for the geometric least squares means ratios (%; combination vs. alone) for analytes' AUCtau, Cmax and Ctau were estimated by a linear mixed effect model and compared to lack of PK alteration boundaries of 70-143%. Safety assessments were conducted during the study. Overall, 95/ 96 subjects completed the study; one Cohort 2 subject withdrew consent. Most adverse events (AEs) were Grade 1 or 2. Most commonly reported AEs were ocular icterus with atazanavir (22%, N=21; Cohort 1 and 3), headache (19%, N=18; all cohorts), and nausea (18%, N=17; all cohorts). One SAE of abdominal pain (Grade 3) was concluded related to ATV/r + TVD by the investigator. Modest increases in LDV and GS-331007 with ATV/r + TVD and a small reduction in SOF with DRV/r + TVD were observed. Increases in ATV and RTV were also observed, and TFV exposures were elevated with both ARV regimens, following either simultaneous or staggered administration of LDV/SOF. The most important findings are summarized in figure 8.
Figure 8: Effect of LDV/SOF on tenofovir pharmacokinetics
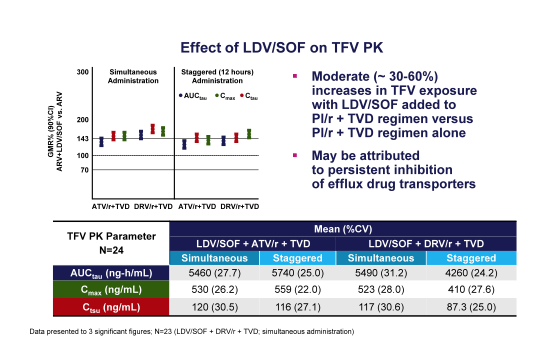
In summary, LDV/SOF increases TFV exposure within RTV-boosted ATV- or DRV-based regimens. Staggered administration seems to have no impact on magnitude of this tenofovir level increase. The safety of higher TFV concentrations in this setting has not been established. Therefore, it appears most appropriate to consider alternative antiretroviral therapies where possible to avoid increases in TFV. Clearly, pre-existing HIV resistance needs to be considered when switching or modifying HIV therapy. Patients which continue on TDF + boosted PI therapy should be monitored carefully for TFV-associated adverse events. [from Jules: a study with TAF in enrolling to see if TAF has better outcome than TDF.]
CROI: Drug Interactions Between the anti-HCV Regimen Ledipasvir/Sofosbuvir and Ritonavir Boosted Protease Inhibitors plus Emtricitabine/Tenofovir DF - (02/23/15)
Another important PK study evaluated the safety and pharmacokinetic (PK) interactions of grazoprevir and elbasvir (Grazoprevir (MK-5172) is a potent, once-daily inhibitor of the hepatitis C virus (HCV) NS3/4A protease and elbasvir (MK-8742) is a potent, once-daily HCV NS5A replication complex inhibitor that are currently being developed as a fixed-dose combination therapy for HCV therapy) when coadministered with dolutegravir (DTG), a HIV-1 integrase strand transfer inhibitor in healthy volunteers (14, poster). Grazoprevir + elbasvir had no clinically meaningful effect on dolutegravir PK, with AUC0-∞ and C24 geometric mean ratios (GMRs) [90% confidence intervals (CIs)] of 1.16 [1.00, 1.34] and 1.14 [0.95, 1.36], respectively. Similarly, elbasvir PK were not affected by coadministration of dolutegravir. However, dolutegravir did result in decreased grazoprevir PK with AUC0-24, Cmax, and C24 GMRs (90% CIs) of 0.81 (0.67, 0.97), 0.64 (0.44, 0.93), and 0.86 (0.79, 0.93), respectively. The mechanism leading to this decrease is currently unclear. However, the lower exposure is within the therapeutic window currently defined for GZR based on available data. These results suggest that no dose adjustments in grazoprevir, elbasvir, or dolutegravir are needed for coadministration in coinfected patients.
What is the role of viral load monitoring under HCV therapy?
Interesting data was presented from the NIAID SYNERGY and ERADICATE study with regard to different HCV viral load assays used throughout the trials and the implication of the lower than the limit of quantification result but still target detectable (15). In two NIAID clinical trials, SYNERGY A (HCV mono-infected, n=17) and ERADICATE (HIV/HCV co-infected, ARV naïve n=13, on cART n=37), subjects were treated with a fixed dose combination of ledipasvir (90 mg) and sofosbuvir (400 mg) for 12 weeks. In both trials, patients were treatment-naïve, non-cirrhotics, and infected with HCV genotype 1. Serial measurements of plasma HCV RNA were performed by the Roche COBAS TaqMan HCV test v1.0 and the Abbott real-time PCR assay. The positive predictive value and negative predictive value at week 4 and end of treatment (EOT) for both assays were calculated. More patients were still detectable with the Abbott assay than with the Roche assay at EOT. Contrary to past experience with interferon-containing treatments though, the presence of detectable HCV RNA levels at EOT was not predictive of relapse in these studies. Also the low negative predictive values at week 4 underscore the importance of continued therapy for patients who fail to achieve undetectable levels of HCV RNA early on during treatment because their chances of achieving SVR are still high. Clearly this raises the question whether viral load monitoring in 12 week HCV treatment cycles is really needed. In Germany most physicians however continue to measure HCV viral load response at week 4 to document adherence but also to help enhance patient motivation by showing the impressive decline in HCV viral load. The amount of patients with HCV RNA ≥ lower limit of quantification (LLOQ) or target detected (TD) < LLOQ at W4 and EOT as well as the respective predictive values are depicted in figure 10.
Figure 10: Patients with HCV RNA ≥ lower limit of quantification (LLOQ) or target detected (TD) < LLOQ at W4 and EOT
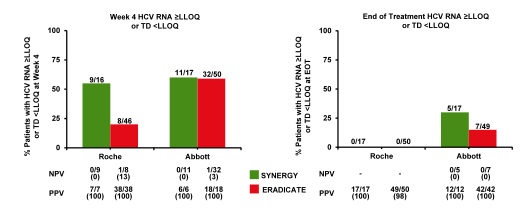
CROI: Utility of Hepatitis C Viral Load Monitoring with Ledipasvir ! and Sofosbuvir Therapy ! (week 4 testing in Harvoni NIH study) - (03/09/15)
Similar findings were reported from a metaanalyses from all Abbvie 3-D studies which assessed HCV RNA viral suppression over time (16). Data from the pooled population of HCV GT-1 mono-infected patients from six phase 3 trials of 3D+/-RBV for 12 weeks or 24 weeks (N=2053) and the HCV/HIV-1 coinfected patients from the Phase 2 TURQUOISE-I trial of 3D+RBV for 12 or 24 weeks (N=63) were included. Patients who experienced non-virologic failure were excluded from the efficacy analysis (N=26 mono-infected; N=1 co-infected). HCV RNA was determined using the Roche COBAS TaqMan RT-PCR assay; lower limit of quantification (LLOQ) =25 IU/ml. SVR12 rates were analyzed according to the first week HCV RNA < LLOQ was attained. SVR12 rates were high in mono- and coinfected patients regardless of time of virological suppression (see figure 11).
Figure 11: SVR12 by Time to HCV RNA Suppression <25 IU/mL
Again these data highlight that time point of achieving undetectability under modern DAA combination therapy has no predictive value for assessing treatment outcome or defining treatment duration at this point.
CROI: HIGH SVR REGARDLESS OF TIME TO SUPPRESSION WITH OMBITASVIR/PARITAPREVIR/R & DASABUVIR + RBV - (02/27/15)
Any other practical news in HCV therapy?
An interesting subanalysis of the TURQUOISE-1 study was presented addressing the question whether ribavirin dose reductions in DAA+RBV combination therapy may potentially impact SVR rates (17, poster). Hemoglobin decreases were comparable between 12- and 24-week treatment arms. Treatment-emergent anemia was reported in 1 patient in the 12-week arm and in 3 patients in the 24-week arm. RBV dose modification due to decreased hemoglobin occurred in 6 patients (4 in the 12-week arm; 2 in the 24-week arm); all achieved SVR12. No patient had a post-baseline Grade 3 or 4 decrease in hemoglobin. No patient used erythropoietin or received a blood transfusion. In summary, among HCV/HIV-1 co-infected treatment-naïve and -experienced patients, anemia events were infrequent with 3D+RBV treatment and did not affect treatment outcomes.
Whats new in acute HCV management?
One of the important remaining questions in the field is the efficacy of DAA based therapy for treatment of acute HCV. Until now only PEG-IFN/RBV has remained the only recommended treatment option for acute HCV in the EASL hepatitis guidelines. At CROI an interim analysis of the first 34 patients treated in the DAHH-Study, evaluating the efficacy and tolerability of a 12-week boceprevir, P+R regimen in AHCV genotype 1 infections in 10 participating Dutch hospitals was presented (18, poster). HIV infected patients visiting the outpatient clinic with a new ALT rise above the upper limit of normal were screened for the presence of HCV RNA. If positive, stored historical plasma samples (within 6 months) were tested to prove that the HCV infection was recent. Boceprevir, pegylated interferon + ribavirin for 12 weeks was started without a PEG-IFN + ribavirin lead-in and was initiated no later than 26 weeks after the presumed day of infection. Primary endpoint was sustained viral response at week 24(SVR24) in patients with no HCV RNA detected (Roche, CAP/CTM) at week4 (RVR4) and in all patients included (secondary endpoint). The study is ongoing but fully enrolled with 65 patients included of which 8 cleared HCV spontaneously and 57 started therapy. 34 patients have reached the SVR12 evaluation endpoint. SVR rates for those 34 patients are shown below in figure 12.
Figure 12: Virological response in ITT patients
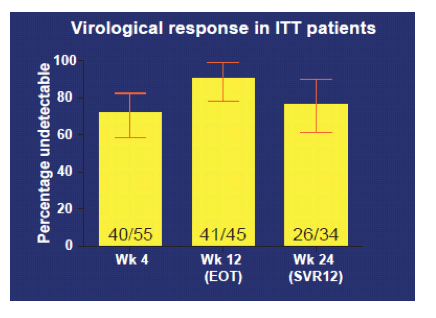
The main findings of the study are that the addition of boceprevir to PEG-IFN + RBV halves therapy duration of acute HCV to 12 weeks and that a high 95% SVR rate was obtained in patients who achieved a RVR4 (95% CI 75%-99%). Overall, SVR24 however was disappointing low with 76% in the ITT population (95% CI 58%-89%). Neither IL28 RS1297 nor the RS809 genotype had an impact on SVR12 results. No unexpected side effects were observed. As not all the patients responded to therapy the role of DAAs in acute HCV still needs to be defined.
After all, in the original N Engl J paper on treatment of acute HCV with non-modified interferon 98% of patients received SVR with interferon alone (19).
Obviously, with advent of DAA therapy the role of PEG-IFN/RBV for treatment of acute HCV infection is going to change. In a poster presentation at CROI potential changes in the annual rates of treatment initiation in Europe for AHC coinfection in the DAA era were presented from the PROBE-C study (20, poster). The PROBE-C study is an observational European cohort on AHC in HIV coinfection. From 2007-2014 483 AHC episodes were documented in 461 HIV-infected patients from Austria, Denmark, France, Germany, Great Britain and Spain. All patients were male, median age was 41 years. Main routes of transmission were MSM (99.4%) and IVDU (0.6%). 77.2% of patients were infected with HCV GT1 and 18.9% with GT4. Median baseline HCV-RNA was 2.190.460IU/mL and median CD4+ T cell count 466 cells/μL. 74,4% of all patients received cART, 86% had baseline suppressed HIV-RNA (In 62/509 (12.2%) episodes AHC resolved spontaneously, in 331/509 (65%) treatment with pegIFN/RBV was initiated within 24 weeks of AHC diagnosis. Median time from diagnosis to treatment initiation was 8 weeks. SVR rate was 70.1%.
Overall, as shown in figure 13 there was an increase in treatment initiation for AHC from 2007-2012. However, since 2012 an annual decline from 71% to 55% was observed. There was no association between treatment initiation and HCV transmission risk, HCV GT, HCV RNA levels or baseline ALT.
Figure 13: Annual rates of treatment initiation for new AHC diagnoses from 2007 to 2014
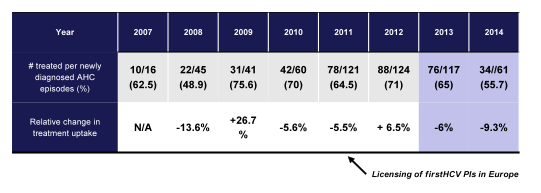
In conclusion, treatment uptake for acute HCV has substantially decreased in the last 2 years potentially reflecting patients' and/or physicians' wish for a short, well-tolerated and highly successful DAA-based therapy which to date is only approved and reimbursed for treatment of chronic HCV infection. However, when treatment during AHC is withheld, more patients remain viremic, which then may foster the epidemic of AHC among HIV-infected MSM. Therefore, studies evaluating safety and efficacy of IFN-free DAA regimens for AHC are urgently needed.
Any data from HCV monoinfection studies?
Updated presentations from Unity 1 and 2 were presented at CROI (21 poster, 22 poster). The all-oral combination of daclatasvir (DCV; pangenotypic NS5A inhibitor), asunaprevir (ASV; NS3 protease inhibitor), and beclabuvir (BCV; non-nucleoside NS5B inhibitor) in a twice-daily fixed-dose combination tablet-DCV-TRIO regimen-was evaluated without ribavirin in HCV genotype (GT) 1-infected treatment-naive and -experienced patients without cirrhosis in a phase 3, open-label, international clinical trial (21). All patients received 12 weeks of treatment with the fixed-dose combination of DCV 30 mg, ASV 200 mg, and BCV 75 mg twice daily. Key efficacy outcomes were SVR12 rates in the treatment-naive and -experienced cohorts, assessed separately. Baseline characteristics in the treatment-naive (N=312) and treatment-experienced (N=103) cohorts were comparable. Overall, patients were 58% male and 26% IL28B (rs1297860) CC genotype; 73% were infected with GT 1a and 27% with GT 1b. SVR12 was achieved by 92% and 89% of treatment-naive and -experienced patients, respectively (see figure 14). 17 patients with GT1b infection had NS5A resistance-associated variants (RAVs) at baseline; all 17 achieved SVR12. 25 of the 34 GT1a-infected patients with baseline NS5A RAVs achieved SVR12. Overall, 34 (8%) patients experienced virologic failure, including 21 with post-treatment relapse and 13 with on-treatment virologic failure. Among patients with GT1a infection who experienced failure, RAVs at positions NS5A-Q30, NS3-R155 and/or NS5B-P495 were the most frequently observed variants. One death occurred post-treatment and was considered not related to study treatment. There were 7 serious adverse events, all unrelated, and 3 (10% of patients included headache, fatigue, diarrhea, and nausea.
Figure 14: SVR12 Rates by Patient Population
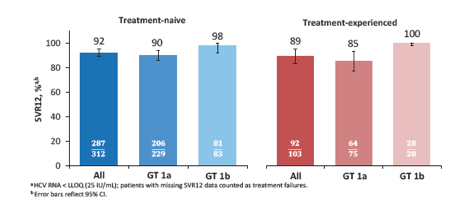
In summary, twelve weeks of all-oral treatment with the fixed-dose combination of DCV, ASV, and BCV achieved high SVR12 rates in 415 patients with chronic HCV GT 1 infection without cirrhosis. Overall cure was higher in genotype 1b than in 1a patients. Whereas all 1b patients seem to respond it becomes more difficult in genotype 1a patients particularly if a history of treatment failure is present. This also raises the question how patients would do under this combination if cirrhotic and treatment experienced with GT1a. DCV-TRIO was well tolerated with low rates of serious AEs (2%) and AE discontinuations (0.7%).
To further study the efficacy and safety of this DCV-TRIO +/- ribavirin in cirrhosis UNITY-2 was designed (22). The all-oral DCV-TRIO regimen including daclatasvir, asunaprevir and beclabuvir (BCV; non-nucleoside NS5B inhibitor) in a fixed-dose combination (FDC) was studied with and without ribavirin (RBV), in treatment-naive and -experienced patients with HCV genotype (GT) 1 infection and compensated cirrhosis in a randomized, phase 3 clinical trial. Patients were randomly assigned to receive a twice-daily FDC of DCV 30 mg, ASV 200 mg, and BCV 75 mg, with blinded RBV or placebo, for 12 weeks. SVR12 rates in the treatment-naive and experienced cohorts were evaluated separately as key efficacy outcomes. Baseline characteristics were comparable between treatment-naive (N=112) and treatment-experienced (N=90) patients. Overall, patients were 66% male and 27% IL28B (rs1297860) CC genotype; 74% of patients had GT1a infection and 26% had GT1b. SVR12 was achieved by 98% and 93% of treatment-naive patients treated with DCV-TRIO + RBV and DCV-TRIO, respectively. Corresponding SVR12 rates for the experienced cohort were 93% and 87%. The SVR rates are shown below in figure 15.
Figure 15: Virologic Response : SVR12 (mITT)
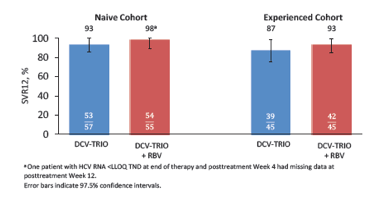
The presence of NS5A or NS3 polymorphisms at baseline did not appear to impact SVR12. Virologic failure was observed in 13 (6%) patients, including post-treatment relapse (10/13) and on-treatment failure (3/13). Ten of 13 patients with virologic failure had both NS5A and NS3 resistance variants at failure. In both groups slightly higher response rates were observed with the addition of ribavirin. Clearly, the question is whether ribavirin is needed for all patients and which impact the genotype 1 subtype may have. The corresponding SVR rates by subtype are shown in Figure 16.
Figure 16: Virologic Response - SVR12 by GT 1 Subtype
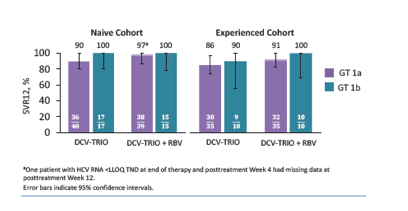
Despite low numbers the data suggests that genotype 1b patients even with cirrhosis continue to show great treatment results, in the naïve cirrhotics even without ribavirin. In the genotype 1a patients however the addition of ribavirin seems to prevent relapse.
Three serious adverse events (SAEs) were considered related to treatment, and there was 1 DCV-TRIO discontinuation due to adverse events.Adverse events reported in >10% of patients included fatigue, headache, nausea, diarrhea, insomnia and pruritus.
In summary, treatment with the DCV/ASV/BCV fixed-dose combination (DCV-TRIO), with or without RBV, in patients with GT 1 and compensated cirrhosis achieved high SVR12 rates after 12 weeks of treatment. SVR12 rates were higher slightly higher for GT1b versus 1a. SVR12 rates were comparable with respect to gender, age, race, baseline HCV RNA, and IL28B genotype. The addition of RBV decreased relapse frequency in GT 1a.
Any other interesting news?
One of the important features of curing HCV through antiviral therapy is the possibility of a HCV reinfection. Obviously, this is of particular concern in HCV patients where no change in transmission risk factors occurs. At CROI an analysis was presented which aimed at estimating the 5-year risk of HCV late relapse or re-infection post-SVR, by risk group (23). The MEDLINE and EMBASE databases were searched for studies analysing late relapse or re-infection post-SVR (typically 24 weeks post-treatment, using pegylated interferon/ribavirin). All studies with >6 months of follow up post-SVR were included. Three groups of patients were analysed: 1. Mono-HCV infected "low risk" patients; 2. Mono-HCV infected "high risk" patients (IV drug users or prisoners); 3. HIV/HCV co-infected patients. Studies of liver transplant patients were excluded. Recurrence was defined as confirmed HCV RNA detectability after SVR (at least 6 months after end of treatment). Results were available from 49 studies in 8534 patients. In each risk group, there were progressive rises in the risk of HCV recurrence with longer follow up time. The corresponding five-year rate (95%CI) of recurrence post-SVR by Risk Group is depicted below in figure 17.
Figure 17: Five-Year Rate (95%CI) of Recurrence Post-SVR, by Risk Group
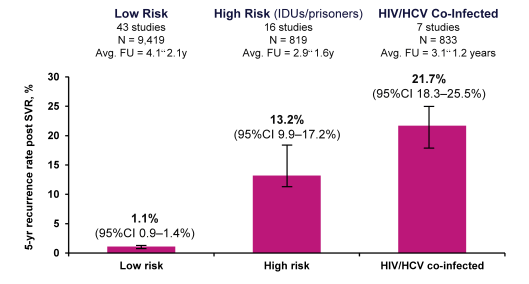
These results clearly underline that drug treatment alone for HCV is not sufficient to control HCV. In addition to HCV therapy harm reduction measures such as syringe exchange, providing clean needles, counseling etc are of utmost importance. For the ongoing epidemic in HIV seropositive MSM psychological, psychotherapeutic and behavioral interventions are needed to impact the high numbers of reinfections through high risk sexual practices with increased blood-blood contacts.
CROI: Risk of late relapse or re-infection with Hepatitis C after Sustained Virological Response: meta-analysis of 66 studies in 11,071 patients - (03/11/15)
One further important topic was the HCV treatment cascade and how the new successful HCV therapies may change the HCV epidemiology and disease burden world-wide. Obviously, the development of new HCV drugs is only one part of the story to control or even eradicate HCV globally; the other side is to increase diagnosis rates and to ensure treatment access. In a study from Switzerland the impact of different treatment strategies on liver fibrosis progression among HIV-infected patients with incident HCV infection was modelled (24, webcast,). An individual-based model of liver disease progression was developed which was parameterized with observed data on incident HCV infections among men who have sex with men from the Swiss HIV Cohort Study (SHCS) and with published data. Patients from HCV infection were simulated through various stages of liver disease: from fibrosis grade F0 to F4, decompensated cirrhosis, hepatocellular carcinoma and death. Liver disease progression was affected by age at HCV infection and alcohol consumption. Patients also progressed through the care cascade: they could be diagnosed, treated and succeed or fail treatment.
Treatment efficacy with Interferon (IFN)-free regimen was assumed to be 90%. Successfully treated patients had a residual liver fibrosis progression of 0.1 times the rate of patients with detectable HCV. Liver-related events and duration of infectiousness (ie. detectable viral load) were compared between the following strategies: treatment of all patients one month after diagnosis, one year after diagnosis or as they reach F2, F3 or F4. Delaying treatment until 1 year after diagnosis or until F2, F3 or F4 led to 14, 43, 142 and 418 additional cases of liver-related deaths per 1000 HCV infections as compared with treating all patients one month after diagnosis. The average time people were infectious increased from 5 years with early (one month after diagnosis) to 21 years with late (F4) treatment. This model suggests that timely treatment of HCV infection is important: Patients can progress to end-stage liver disease after HCV clearance if treatment is delayed until later stages of liver disease due to imperfect treatment responses and residual fibrosis progression in HIV-infected patients. Delaying treatment also increases the risk of HCV transmission: the average time during which patients are infectious is four times higher if patients are treated in F4 than if they are treated one month after diagnosis. Clearly these results help to stimulate the discussion whether guidelines recommended prioritization of HCV treatment only in advanced fibrosis (F3, F4) makes sense or whether earlier treatment could not only prevent progression to end-stage liver disease and associated complications but also prevent HCV transmission to others.
Summary
· Ledipasvir/sofosbuvir for 12 weeks achieves impressive SVR rates of 96% which are very similar to treatment outcome reports from studies with this DAA combination in HCV mono-infection. Patients on tenofovir need careful renal monitoring as ledipasvir may increase tenofovir exposure. Overall, tolerability of this regimen was very good with no treatment related discontinuations.
· Daclatasvir and sofosbuvir for 12 weeks likewise achieved very high HCV cure rates of 96% in HIV/HCV coinfected subjects and predominantly HCV GT1 infection. Tolerability of this regimen was very good with no treatment related discontinuations.
· In HIV/HCV GT1/4 null responders, one third of whom had cirrhosis, a 24-week regimen combining daclatasvir/asunaprevir/PR was associated with a very high SVR12 rate. The relatively high rate of SAEs underlines why IFN-free regimes are clearly the way forward particularly in cirrhosis.
· Cohort data from real-life patient with a substantial proportion of patients with advanced fibrosis liver cirrhosis and high rates of previous treatment failure respond well to sofosbuvir containing regimens. Cirrhosis but not HCV/HIV-coinfection seems to be associated with a lower SVR.
· SVR12 rates in real-life cohorts are completely comparable between co- and HCV mono-infected subjects.
· SOF+SMV+/-RBV treatment may be appropriate for carefully selected HCV PI-experienced G1 pts including those with cirrhosis and as predicted by genotypic resistance testing.
· LDV/SOF increases TFV exposure within RTV-boosted ATV- or DRV-based regimens. Staggered administration seems to have no impact on magnitude of this tenofovir level increase. The safety of higher TFV concentrations in this setting has not been established. Therefore, it appears most appropriate to consider alternative antiretroviral therapies where possible to avoid increases in TFV. Clearly, pre-existing HIV resistance needs to be considered when switching or modifying HIV therapy. Patients which continue on TDF + boosted PI therapy should be monitored carefully for TFV-associated adverse events.
· No dose adjustments in grazoprevir, elbasvir, or dolutegravir are needed for coadministration in coinfected patients.
· The time point of achieving undetectability under modern DAA combination therapy has no predictive value for assessing treatment outcome or defining treatment duration at this point.
· Treatment uptake for acute HCV has substantially decreased in the last 2 years potentially reflecting patients' and/or physicians' wish for a short, well-tolerated and highly successful DAA-based therapy which to date is only approved and reimbursed for treatment of chronic HCV infection. However, when treatment during AHC is withheld, more patients remain viremic, which then may foster the epidemic of AHC among HIV-infected MSM. Therefore, studies evaluating safety and efficacy of IFN-free DAA regimens for AHC are urgently needed.
· Treatment with the DCV/ASV/BCV fixed-dose combination (DCV-TRIO), with or without RBV, in patients with GT 1 and compensated cirrhosis achieved high SVR12 rates after 12 weeks of treatment. SVR12 rates were higher slightly higher for GT1b versus 1a. SVR12 rates were comparable with respect to gender, age, race, baseline HCV RNA, and IL28B genotype. The addition of RBV decreased relapse frequency in GT 1a.
· Timely treatment of HCV infection is important: Patients can progress to end-stage liver disease after HCV clearance if treatment is delayed until later stages of liver disease due to imperfect treatment responses and residual fibrosis progression in HIV-infected patients. Delaying treatment also increases the risk of HCV transmission.
References
1. Naggie S, et al.: Ledipasvir/Sofosbuvir for 12 Weeks in Patients Coinfected With HCV and HIV-1. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 152LB
2. Wyles D, et al.: Daclatasvir in Combination with Sofosbuvir for HIV/HCV Coinfection: ALLY-2 Study. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 151LB
3. Eley T, et al.: Daclatasvir: Overview of Drug-Drug Interactions With Antiretroviral Agents and Other Common Concomitant Drugs. HIV DART 2014, December 9-12, 2014
4. Piroth L, et al.: High Efficacy of Daclatasvir/Asunaprevir/PR in HIV/HCV1-4 Null Responders (ANRS HC30) 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 146
5. Christensen C et al.: German Cohort on Sofosbuvir-Based Therapy for HIV/HCV and HCV Infection (GECOSO). 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 646
6. Del Bello D, et al.: Real-World Data on HIV-Positive Patients With HCV Treated With Sofosbuvir and/or Simeprevir. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 647
7. Perumalswami PV et al,: Simeprevir and Sofosbuvir Regimens for Hepatitis C: Decompensation and Serious AEs. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 648
8. Grant JL, et al.: Successful HCV Treatment with Direct Acting Antivirals in HIV/HCV Patients. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 649
9. Gilmore J, et al.: Effectiveness of Sofosbuvir/Simeprevir for HIV/HCV Patients in Clinical Practice. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 645
10. Marks K, et al.; Sofosbuvir, Simeprevir, +/- Ribavirin in HCV Protease Inhibitor-Experienced Patients. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 644
11. Cope R, et al.: Majority of HIV/HCV Patients Need to Switch ART to Accommodate Simeprevir. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 651
12. Naqvi A, et al.: Sofosbuvir/Daclatasvir in HIV/HCV Co-infected Patients with Extensive Liver Fibrosis. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 650
13. German P, et al.: Drug-Drug Interactions between Anti-HCV Regimen Ledipasvir/Sofosbuvir and Antiretrovirals. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 820
14. Yeh WY, et al,: Drug-Drug Interaction Between HCV Inhibitors Grazoprevir/Elbasvir With Dolutegravir. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 522
15. Sidharthan S, et al.: Utility of Hepatitis C Viral-Load Monitoring With Ledipasvir and Sofosbuvir Therapy. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 689
16. Wyles D, et al.: High SVR regardless of Time to Suppression with ABT-450/r/Ombitasvir & Dasabuvir+RBV. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 147
17. Sulkowski M, et al.: Hematologic Analysis of ABT-450/r/Ombitasvir and Dasabuvir + RBV in TURQUOISE-I.
18. Hullegie S, et al.: SVR12 Results After 12w Boceprevir + P/R in the Dutch Acute Hepatitis C in HIV Study. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 669
19. Jaeckel E, et al.: German Acute Hepatitis C Therapy Group. Treatment of acute hepatitis C with interferon alfa-2b. N Engl J Med. 2001;345:1452-7.
20. Boesecke C, et al.: Does the Availability of New DAAs Influence Treatment Uptake in Acute Hepatitis C in HIV Coinfection? 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 670
21. Poordad F, et al.: UNITY-1: Daclatasvir/Asunaprevir/Beclabuvir for HCV Genotype 1 Without Cirrhosis. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 687
22. Muir A, et al.: UNITY-2: Daclatasvir/Asunaprevir/Beclabuvir ± RBV for HCV Genotype 1 With Cirrhosis. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 688
23. Hill A, et al.: Five-Year Risk of Late Relapse or Reinfection with Hepatitis C after Sustained Virological Response: Meta-analysis of 49 Studies in 8534 Patients. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 654
24. Zahnd et al.: Impact of Deferring HCV Treatment on Liver-Related Events in HIV+ Patients. 22nd Conference on Retroviruses and Opportunistic Infections, February 23-26, 2015; abstract 150
|
| |
|
|
|
|
|